EJCRIM 2023 CiteScore
| 2.1 = | 1.730 Cit. to date |
| 842 Docs. to date |
Last updated on 05 April, 2024
Updated monthly
Updated monthly
Powered by



|
Views: 112
HTML: 7
PDF: 34
|
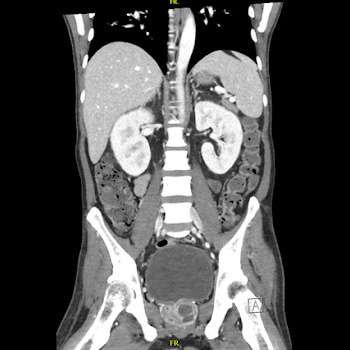
Background: Melioidosis is an infection caused by Burkholderia pseudomallei, a Gram-negative bacterium. It is a disease endemic to Southeast Asia and northern Australia although its global incidence has been rising. It most commonly infects people with certain identified risk factors such as diabetes, alcoholism, thalassemia, and underlying chronic disease involving lungs, kidney and liver. This bacterium is capable of producing a wide array of clinical manifestations ranging from asymptomatic disease to localised infections such as in the lung, bone or skin to disseminated infection.
Case description: This is a case, from United Arab Emirates, of a 40-year-old male recently diagnosed with diabetes who presented with multiple abscesses and was eventually diagnosed with disseminated melioidosis. He was treated successfully with antibiotics and drainage of abscesses.
Conclusion: In non-endemic regions, melioidosis can be easily missed in common diagnostic approaches. This gap of awareness could delay the diagnosis and allow further deterioration of the patient due to complications. Thus, case reports like this can enlighten internists about changing incidences and complexity of clinical presentations, thus preparing them to better handle such patients in the future.
|
Views: 19
HTML: 0
PDF: 4
|
Introduction: EML4-ALK is an oncogenic driver, seen in around five per cent of advanced non-small-cell lung cancer (NSCLC) patients, which can be targeted with anaplastic lymphoma kinase tyrosine kinase inhibitors with great response rates. Disease flare refers to sudden rapid disease worsening on tyrosine kinase inhibitors (TKI) discontinuation, which is associated with shorter survival and worse outcomes. Here, we review cases previously published in the literature where patients developed disease flares, and contrast this with our patients who had prolonged survival despite TKI discontinuation.
Case description: We report three different patients with advanced ALK-positive NSCLC seen at our institute, who had EML4-ALK translocation variant 1 oncogenic driver on next-generation sequencing. They received treatment with several different ALK inhibitors before opting to discontinue TKI. They were able to come off TKI safely without developing disease flare and had prolonged survival.
Discussion: Shorter time to progression on TKI, presence of symptoms with disease progression or central nervous system/pleural metastasis have been previously linked with development of flare, although this was not seen in our case series. Tumour response at the time of treatment discontinuation, line of therapy, overall disease burden, fusion variant and co-alteration status can affect the prognosis of these patients after ALK TKI cessation. In particular, variant 1 and wild-type TP53 status may be a suitable patient population for dose optimisation strategies. Intermittent TKI dosing strategies may help to avoid acquiring resistance mutations and prevent long-term treatment toxicities.
Conclusion: It is important for clinicians to identify patients at risk for developing disease flare on TKI discontinuation to improve outcomes. Intermittent TKI dosing strategies require further investigation.

|
Views: 17
HTML: 0
PDF: 6
|
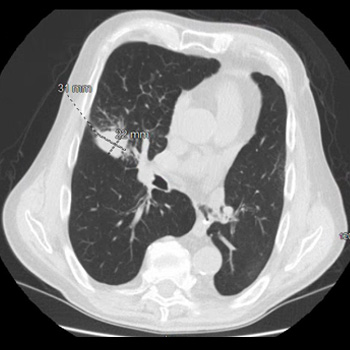
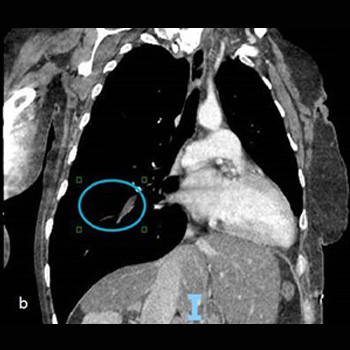
Background: Small cell lung cancer is an aggressive tumor with a poor prognosis that requires prompt treatment. While radiotherapy may enhance survival when superior vena cava syndrome is present, radiation therapy–induced pericardial disease can be a potential complication.
Case Report: A 55-year-old man, who recently underwent radiotherapy for stage IV small-cell lung cancer complicated by superior vena cava syndrome, presented with chest pain and dyspnea. In the emergency room, he was dyspneic, hypotensive, and tachycardic. Pulmonary auscultation revealed the absence of lung sounds on the right. The initial electrocardiogram showed ST-segment elevation in lateral leads and in lead DII, with reciprocal changes in lead DIII. A bedside transthoracic echocardiogram revealed cardiac tamponade and emergent pericardiocentesis was performed, removing 500 ml of purulent fluid, resulting in an immediate clinical improvement. Thoracentesis was also performed, showing no empyema. Large spectrum empirical antibiotic therapy was started. Cultures from the pericardial fluid and peripheral blood grew multi-sensitive Streptococcus pneumoniae. Cytological analysis of the pericardial fluid was consistent with infection. The patient improved after 2 weeks of targeted antibiotic therapy and underwent the first cycle of chemotherapy. He was discharged with an early scheduled pulmonology appointment.
Conclusions: Although the most common causes of pericardial effusion in lung cancer are malignant, non-malignant etiologies should also be considered. This patient had an infectious pericardial effusion most probably due to a pericardial-mediastinal mass fistula caused by radiotherapy. This was a diagnostic challenge, both in the emergency room as well in the inpatient setting.
|
Views: 20
HTML: 0
PDF: 7
|
Background: Fournier’s gangrene represents a life-threatening necrotising infection affecting the perineal region, while hidradenitis suppurativa is characterised by a chronic inflammatory skin condition. The simultaneous occurrence of both conditions is exceedingly rare.
Case description: A 42-year-old female with a documented history of severe untreated hidradenitis suppurativa presented for shortness of breath, fever and lethargy, along with extensive wounds and skin breakdown involving the left axilla, perineum, lower back, lumbosacral region and bilateral gluteal areas, extending to the perineum. Upon presentation, the patient was in a state of septic shock, and a diagnosis of actively manifesting Fournier’s gangrene was established at the site of the pre-existing hidradenitis suppurativa lesions. Despite the implementation of an aggressive multidisciplinary approach incorporating surgical interventions, antibiotic therapy and intensive care measures, the patient’s condition deteriorated, culminating in septic shock, multi-organ failure and eventual demise. In this report, we discuss both clinical entities, their similarities and differences, and the possible mechanisms by which they may have co-occurred.
Conclusion: The co-existence of hidradenitis suppurativa and Fournier’s gangrene poses unique challenges, given the rapid progression of Fournier’s gangrene within the context of hidradenitis suppurativa, potentially suggesting the latter as a predisposing factor. This case underscores the importance of vigilant screening and management of hidradenitis suppurativa.
|
Views: 22
PDF: 19
HTML: 2
|
Background: Studies have shown major cardiovascular effects associated with ketamine use disorder including dose-dependent negative inotropic effects. Preoperative ketamine use has been linked to ketamine-induced stress cardiomyopathy.
Case presentation: A 28-year-old female with a history of recurrent cystitis and ketamine use disorder (twice weekly for 14 years) presented with bilateral lower extremity oedema and shortness of breath for 3 months. She was tachycardic with a troponin level of 0.07 ng/ml and a B-type natriuretic peptide (BNP) level of 2511 pg/ml. Electrocardiogram showed normal sinus rhythm and transthoracic echocardiography (TTE) showed left ventricular ejection fraction (EF) of 15%, dilated left ventricle, and severe tricuspid and mitral regurgitation. Computed tomography (CT) scan of the chest and abdomen showed bilateral pleural effusions with congestive hepatopathy and ascites. The patient was started on intravenous furosemide, metoprolol, and sacubitril/valsartan. Rheumatological workup including complement levels, and antinuclear anti-double-stranded DNA was negative. A repeat TTE 2 weeks later revealed an EF of 25% and moderate tricuspid regurgitation. Four months later, the EF was 54% with normal left ventricular cavity size.
Conclusion: Although ketamine use disorder is increasing, data on long-term side effects is minimal. Screening for ketamine use disorders should be considered in patients presenting with acute systolic heart failure. Long-term studies are needed to evaluate the benefits of adding ketamine screening to standard urine toxicology.
|
Views: 22
PDF: 9
HTML: 2
|
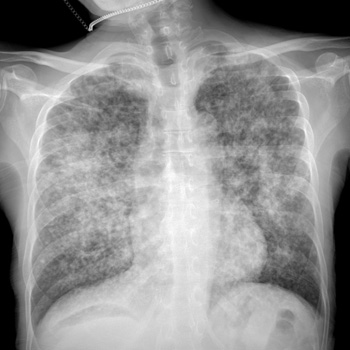
Prostate cancer can metastasise to the lung. Most common presentations described in the literature are solitary pulmonary nodules, lymphangitic spread and, rarely, pleural effusion.
We describe a case of prostate adenocarcinoma with diffuse bilateral reticulonodular and lymphangitic pulmonary metastasis, and malignant pleural effusion while being on androgen deprivation therapy.
|
Views: 26
HTML: 0
PDF: 10
|
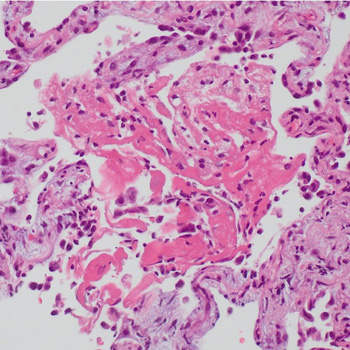
A patient initially treated with corticosteroids for cryptogenic organising pneumonia following pulmonary infarction, developed a worsening condition with progressive cavitary formations in both lower lung lobes. Contrast-enhanced chest computed tomography revealed a pulmonary embolism, and serum anti-Aspergillus IgG antibody analysis yielded a strong positive result. Consequently, the patient was diagnosed with pulmonary infarction with Aspergillus infection; organising pneumonia in surrounding areas reflected the repair process. Following treatment with anticoagulants and antifungal agents, the patient was successfully discharged. Hence, pulmonary infarction should be considered in cases of refractory lung lesions.
|
Views: 53
HTML: 4
PDF: 15
|
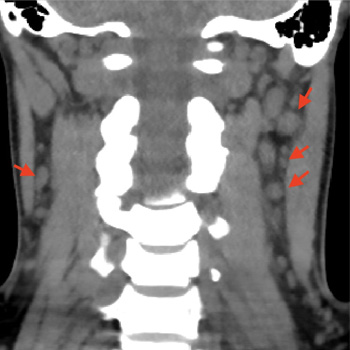
Syphilis, a disease caused by the bacteria Treponema pallidum, has a multitude of clinical manifestations and is classified into primary syphilis, secondary syphilis and tertiary syphilis, based on clinical presentations and the time elapsed since the primary infection. The secondary stage of the disease can affect multiple organs and systems, and some of these involvements may be general and non-specific, justifying its name as ‘the great imitator’. We present a case of a 30-year-old woman with a history of painful neck lymph nodes with progressive enlargement, persistent headache, weight loss, myalgia and alopecia. During investigations, stomatitis on the dorsal face of the tongue developed. A secondary study showed serum positive for rapid plasma reagin (RPR) and T. pallidum haemagglutination (TPHA), negative RPR in cerebrospinal fluid and normal MRI, thus the diagnosis of secondary syphilis was made. The patient was treated with a single dose of penicillin with complete resolution of symptoms. The case highlights the need for an exhaustive clinical examination, especially in cases presenting with non-specific and general symptoms, and raises awareness for this disease which has increased its prevalence in the last decades.
|
Views: 44
HTML: 4
PDF: 22
|
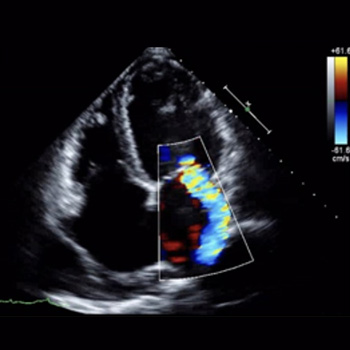
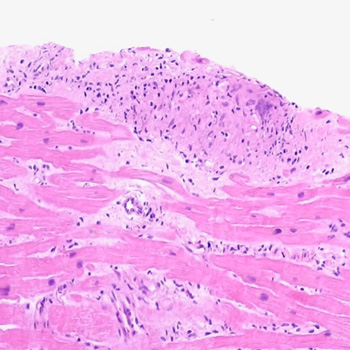
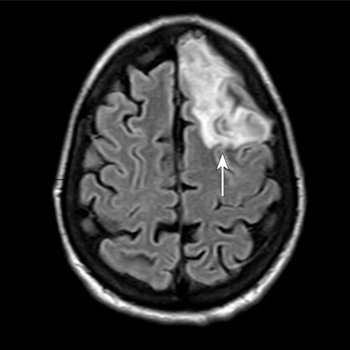
Introduction: Sarcoidosis has many possible clinical presentations since it can affect any organ, most commonly the lungs. The hallmark of the disease consists of the formation of non-necrotising granulomas. Pathogenesis is thought to rely on the interplay of genetic, environmental and epigenetic factors. This case highlights the importance of a thorough clinical history and physical examination, and the correlation with imaging findings in the diagnostic work-up of the non-ischaemic cardiomyopathy.
Case description: A 57-year-old woman was admitted due to the sudden onset of malaise, dizziness, and chest discomfort. Sustained monomorphic ventricular tachycardia was evidenced and the patient rapidly evolved with haemodynamic instability; she underwent successful electrical cardioversion. The electrocardiogram afterwards showed a high-risk electrocardiographic pattern. Invasive coronary angiography excluded obstructive epicardial coronary lesions. Physical examination revealed skin lesions on the lower limbs which raised suspicion for erythema nodosum and therefore a biopsy was performed. Transthoracic echocardiography and cardiac magnetic resonance imaging revealed features consistent with an inflammatory cardiomyopathy, and an implantable cardioverter-defibrillator was placed. The histologic examination of the cutaneous lesions showed a non-necrotising granulomatous inflammatory process. Radionuclide imaging was inconclusive. The patient underwent an endomyocardial biopsy, which confirmed the diagnosis of systemic sarcoidosis with cardiac involvement.
Conclusions: Systemic sarcoidosis with cardiac involvement is a challenging diagnosis. The role of imaging techniques such as transthoracic echocardiography, cardiac magnetic resonance imaging and radionuclide imaging is essential in raising suspicion and diagnosing this pathology. Endomyocardial biopsy is the ‘gold standard’ for its diagnosis; however, it has a low diagnostic yield.
|
Views: 77
HTML: 31
PDF: 27
|
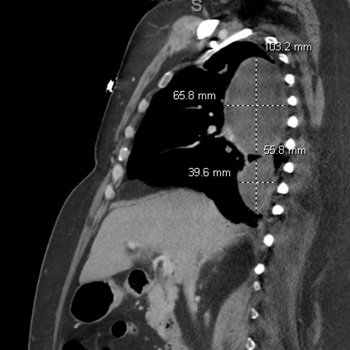
Background: Community-acquired bacterial meningitis in adults represents one of the most severe infectious diseases worldwide with potentially life-threatening medical complications. Several infectious agents can cause acute meningitis. Although group B Streptococcus is more prevalent in newborns, infection can also lead to meningitis in older adults, particularly those with underlying health issues.
Case Description: A 53-year-old woman with a body mass index of 28.7 kg/m2, type 2 diabetes mellitus, and dyslipidaemia presented to the emergency department of Santa Maria della Stella Hospital (Orvieto, Italy) with confusion, low-grade fever, echolalia, and hyperglycaemia. Computed tomography scans of the brain revealed a hypodensity in the left anterior frontal lobe and an osteodural defect of the rhinobase. Meningitis was suspected and empiric broad-spectrum antibiotic therapy with corticosteroids and insulin were administered while the results of the cerebrospinal fluid analysis confirmed the diagnosis of group B Streptococcus meningitis. Repeat imaging at 48 hours revealed enlargement of the hypodense lesion. The frontal assessment battery indicated deficits in executive functions. Prompt treatment led to rapid clinical improvement. Following the restoration of euglycemic status and hemodynamic stabilization, a follow-up magnetic resonance imaging confirmed the ischaemic lesion and showed cerebrospinal fluid in the sella turcica. The patient was then transferred to neurorehabilitation.
Conclusions: The complex interactions among multiple risk factors resulted in an atypical clinical case of group B Streptococcus meningitis, which was promptly treated with empiric antibiotic therapy to mitigate neurocognitive deficits.
|
Views: 11
HTML: 2
PDF: 8
|
Histoplasmosis is a soil based dimorphic fungus endemic to the Midwest and Southeastern United States and is responsible for infection through inhalation of conidia. Infection is usually asymptomatic, as the fungal growth is contained by formation of granulomas. However, dissemination can occur in immunocompromised hosts due to the lack of optimal activity of interferon gamma, tumor necrosis factor-alpha (TNF-alpha) and interleukin-17. There is a significant overlap between the symptomatology of histoplasmosis and granulomatosis with polyangiitis (GPA). We are reporting a case of a 48-year-old female who presented with high grade fever, worsening generalized weakness and tachycardia. She had a previous history of bilateral cavitary lung lesions for which she was evaluated at an outside facility. As her entire infectious work up was negative and found to be positive for antineutrophil cytoplasmic antibody (ANCA), a diagnosis of GPA was made and she was initiated on rituximab infusions 7 weeks prior to her presentation to our facility. Repeat infectious work up at our facility was positive for (1,3) beta-D-glucan test and urine histoplasma antigen. Prompt discontinuation of rituximab and initiation of systemic antifungal therapy led to clinical improvement. Based on this experience we would like to highlight the association of histoplasma with ANCA positivity along with the importance of closely monitoring these patients, for possible clinical worsening after the initiation of immunosuppressive therapy, despite the negative infectious work up.

|
Views: 15
HTML: 1
PDF: 18
|
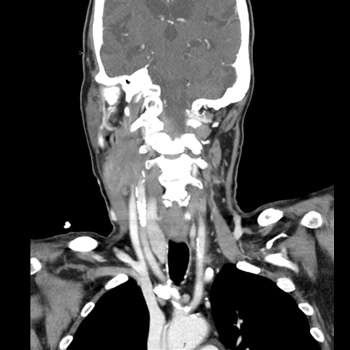
Introduction: Caustic substances ingestion results in a complex syndrome. The patient characteristics and severity of injury are important prognostic predictors. The monitoring of clinical changes and the multidisciplinary approach are necessary to prevent death in the early stages of the poisoning.
Case description: The case report describes the suicide of a woman by ingestion of a large amount of 15% sulfuric acid for suicidal purposes (15–20 ml). The initial conditions were stable, and no changes were found on a CT scan. However, the main sign was a severe metabolic acidosis. After 7 hours, haematemesis and oedema of the larynx appeared, and oro-tracheal intubation and ICU admission were necessary. Consequent progressive haemodynamic deterioration with persistent severe metabolic acidosis, increasing lactates and septic shock appeared. A new CT scan with contrast was performed 22 hours later detecting diffuse perforations and liquid in pleurae and abdomen. A pleural sample showed necrotic liquid. The death was 24 hours after ingestion and no surgical treatment was performed because of the diffuse lesions to the thoracoabdominal districts.
Conclusions: Early detection of gastroenteric lesions and the monitoring of clinical changes are mandatory to avoid the death of the patient. Gastroenteric perforations require an immediate radiological evaluation and surgical treatment. The clinical case report states the severity of prognosis was related to high doses of sulfuric acid ingestion. The immediate metabolic acidosis is related to quick subsequent severe systemic pathological lesions of the gastrointestinal tract. The severity of absorption metabolic acidosis, consequently, may be an early and prognostic sign of the late chest and abdominal lesions.
|
Views: 19
HTML: 0
PDF: 15
|
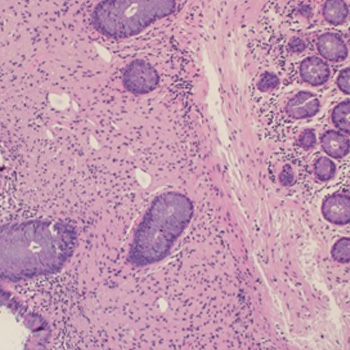
Internal medicine is the specialty with the most semiological training and it is taught that the combination of a complete clinical history with a thorough physical examination allows for a diagnosis to be reached in the majority of cases. We present a clinical case where an incomplete physical examination interfered with the course of hospitalisation. In a growing technological world where complementary diagnostic tests often allow us to see what is impossible to the eye, the physical examination is often neglected.
|
Views: 83
HTML: 13
PDF: 53
|
Schwann cells are found in the peripheral nervous system and can sometimes appear as benign hamartoma lesions in various parts of the body. Although rare in the gastrointestinal (GI) tract, they have been observed in the colon.
Recently, mucosal Schwann cell hamartomas of the GI tract have been studied, and it was discovered that they had yet to be investigated up to 2009. In this context, we present the case of a 60-year-old man who was found to have lesions in the transverse colon during a routine colonoscopy. No further investigations were conducted since these lesions have not been associated with any risk of malignancy transformation and have not been linked to any inherited syndromes.
|
Views: 25
HTML: 3
PDF: 18
|
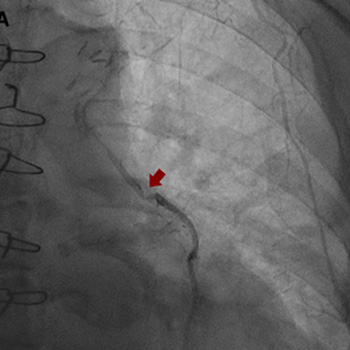
Intracoronary in-stent restenosis (ISR) is a phenomenon that generally occurs between 3 and 6 months after stent placement. With the introduction of drug-eluting stents (DES), the incidence of ISR has decreased but not disappeared. We report a case of reiterant in-stent restenosis of an 81-year-old female patient who underwent multiple percutaneous coronary intervention and two coronary artery bypass surgeries. ISR is possibly associated with extra-stent, stent-related and intra-stent factors. Here, we excluded the first two and focused on the intra-stent factors that seem more likely in our case. A challenging diagnostic workup led us to the hypothesis of a coronary vasculitis potentially triggered by some component of the stent in a predisposed patient carrier of non-disease-specific ANA, with an exaggerated immune response. No recurrence of ISR occurred after the introduction of steroids. Biological and intra-stent causes of ISR should be taken into careful consideration to aim for the early detection of the underlying mechanism of restenosis and to embrace the best therapeutic strategy.
|
Views: 41
HTML: 5
PDF: 30
|
Background: Although there is no specific therapy for COVID-19, it is recommended that patients with severe SARS-CoV-2 infection are treated with corticosteroids and anti-IL-6 receptor monoclonal antibodies. Both COVID-19 itself and the treatment modalities mentioned above have suppressive effects on the immune system which may lead to an increased susceptibility to other infections. In patients with latent tuberculosis (TB) reactivation of TB infection after recovery from severe COVID-19 has been described. Most of these cases have occurred in parts of the world where tuberculosis is endemic.
Case description: The patient is a female in her 70s who was born and raised in Southeast Asia and has lived in the Netherlands for more than 30 years. She was treated for a severe COVID-19 requiring mechanical ventilation for several weeks and pharmaceutical treatment with corticosteroids and anti-IL-6 receptor monoclonal antibodies (Sarilumab). She recovered well. Two years later she was readmitted with symptoms of a serious pulmonary infection and meningitis. Her condition deteriorated in a short time. An active TB infection was diagnosed. Despite adequate antibiotic treatment and supportive therapy her condition worsened and four days after admission to the ICU she deceased.
Discussion: Reactivation of latent TB after recovery from a severe COVID-19 has been described several times and may occur several months after the SARS-CoV-2 infection. In this case the reactivation presented two years after COVID-19. This case illustrates that long-term follow-up of patients with latent TB that recover from a severe COVID-19 may be indicated.
|
Views: 41
PDF: 26
HTML: 19
|
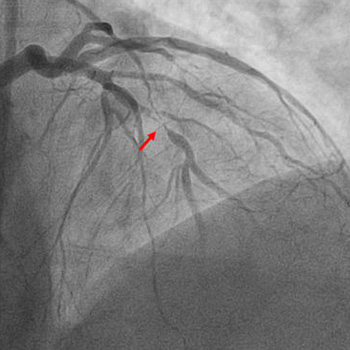
An acute ST-elevation myocardial infarction (STEMI) followed by reinfarction within a short period of time is typically due to stent thrombosis. However, a STEMI caused by occlusion of one vessel followed by a repeat infarction due to occlusion of a different vessel which was seemingly innocent a few hours earlier is extremely rare. We present the case of a 61-year-old male with a past medical history of prediabetes, hyperlipidemia, tobacco use, and gastroesophageal reflux disease who presented to the emergency department with complaints of chest pain. His initial electrocardiogram (EKG) revealed ST elevation in leads II, III and aVF with reciprocal changes in leads I and aVL. He promptly underwent cardiac catheterization and had percutaneous coronary intervention with placement of two drug-eluting stents (DES) in the right coronary artery (RCA). At that time coronary angiography revealed 50% stenosis of the left anterior descending (LAD) artery and 60% stenosis of the second diagonal branch artery. Shortly after the procedure he was asymptomatic, and the post procedure EKG demonstrated resolution of the ST elevations. However, within 2 hours he developed chest pain and was found to have new ST elevations in the anterolateral leads. Repeat cardiac catheterization revealed patent RCA stents with subtotal occlusion of the LAD and another DES was placed. After the second procedure the patient remained hemodynamically stable, EKG changes resolved, and he was kept on eptifibatide infusion for 18 hours after which he was switched to dual antiplatelet therapy and ultimately discharged home.
|
Views: 44
HTML: 2
PDF: 34
|
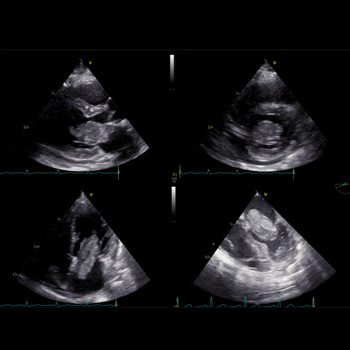
Introduction: Myxoma of the left atrium is a less typical cause of mitral obstruction. If this develops, a flash pulmonary oedema can be the first manifestation.
Case description: We present a case report of a 50-year-old woman who was admitted to our internal department because of dyspnoea. The patient overcame a stroke three years before the index hospitalisation with a negative transthoracic echocardiography. By anamnesis and physical examination, an exacerbation of COPD was assumed, and the patient was treated accordingly. As the patient showed numerous risk factors for heart failure with preserved ejection fraction, transthoracic echocardiography was performed. A large polypoid mass was found in the left atrium, which caused severe mitral obstruction. Subsequent transoesophageal echocardiography confirmed this finding. The patient underwent urgent cardiac surgery, and the tumour was successfully resected. A histological examination revealed a cardiac myxoma. After the cardiac surgery the patient felt well, and no recurrence of the tumour occurred.
Conclusions: We provide a case report of a fast-growing myxoma that was incidentally found in a patient with dyspnoea. We highlight the fast growth rate of the tumour and the potential for misdiagnosed signs of pulmonary oedema caused by mitral obstruction.
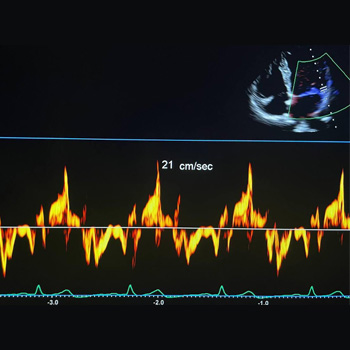
|
Views: 62
HTML: 6
PDF: 32
|
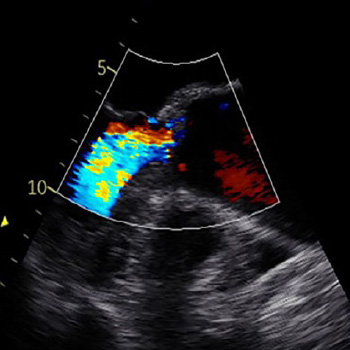
Mitral valve prolapse (MVP) is a primary valvular disease of the mitral valve with a prevalence of 2.4% of the general population. Valve abnormalities range from simple fibroelastic deficiency of the leaflets to diffuse myxomatous degenerative changes. MVP is a usually a benign condition. However, the scattered reports of sudden cardiac death in patients with MVP in the absence of severe mitral insufficiency or coronary artery disease suggest the existence of a malignant phenotype of MVP. We report a case of a young female who survived life-threatening arrhythmias and cardiac arrest and was found to have characteristic features of the malignant phenotype of MVP and had an implantable cardioverter defibrillator as a secondary prevention.
|
Views: 27
HTML: 2
PDF: 29
|
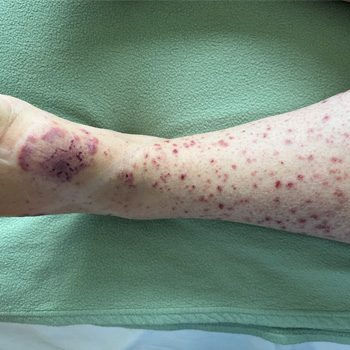
Background: Annular erythema is a rare manifestation of leukocytoclastic vasculitis. It may be associated with various drugs, infections, malignancies, or systemic diseases.
Case description: A 36-year-old woman with no personal medical history presented with annular erythema with target lesions and petechial purpura. The patient had fever and joint arthralgia. A skin biopsy showed leukocytoclastic vasculitis with IgA deposits on direct immunofluorescence. The diagnosis of immunoglobulin A vasculitis with annular leukocytoclastic vasculitis was made. The patient showed global improvement with topical steroids without relapse.
Conclusion: An annular variant of leukocytoclastic vasculitis is a rare manifestation of immunoglobulin A vasculitis.
|
Views: 29
HTML: 2
PDF: 27
|
Case description: We describe a case of a patient treated with pembrolizumab (an immune checkpoint inhibitor) for metastatic scalp melanoma. He had a previous history of colorectal cancer, prostatic cancer and chronic polymyalgia rheumatica. The patient was known to have a stable ascending aortic aneurysm of 4.5 cm. However, he developed a rapid expansion of the ascending aortic aneurysm with the size crossing the threshold for surgery. The patient was referred to the cardiothoracic surgery service for intervention and he subsequently underwent surgery. The patient was electively admitted one week later for resection of aortic aneurysm, aortoplasty and external graft fixation. Pathologically, gross evidence of dissection was not identified; however, the histological analysis of the media showed laminar medial necrosis, multifocal in nature, with occasional clusters of histiocytic cells appreciated at their edge reminiscent of that seen in an inflammatory aortitis (granulomatous/giant cell type).
Discussion: Immune checkpoint inhibitor-induced aortitis is becoming increasingly evident, and its presentation can vary. It has been discovered incidentally on surveillance imaging with the use of nivolumab. In other cases, patients have been symptomatic to severely symptomatic. Atezolizumab with carboplatin and etoposide has been reported to cause abdominal aortitis which was responsive to corticosteroids and subsequent discontinuation of atezolizumab. Pembrolizumab has been linked to a case of transverse aortic arch aortitis. In our case, the inflammatory aortitis due to pembrolizumab was the cause of the rapid expansion of the ascending aortic aneurysm.
Conclusion: Patients with known aortic aneurysms should undergo careful surveillance when commencing immune-checkpoint inhibitor therapy.
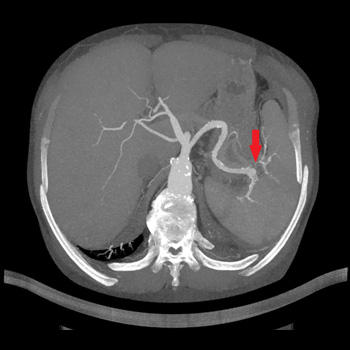
|
Views: 24
HTML: 0
PDF: 9
|
Introduction: Orthotopic heart transplantation is the gold standard for the treatment of advanced heart failure in the absence of contraindications. Infective endocarditis is a rare complication in patients after heart transplantation. The treatment of endocarditis after heart transplantation is challenging since there is a need for ongoing immunosuppression.
Case description: We present the case of a 51-year-old orthotopic heart transplant recipient enrolled in a chronic dialysis program, in whom we diagnosed and successfully treated recurrent infective endocarditis of the mitral valve caused by Enterococcus and Enterobacter species. Despite the complicated course of the disease, the treatment was successful.
Conclusions: Recurrent infective endocarditis after heart transplantation can be treated successfully with a multidisciplinary approach and robust antimicrobial therapy.
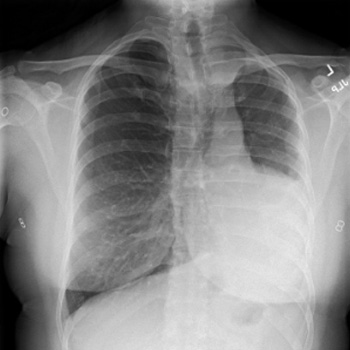
|
Views: 31
HTML: 6
PDF: 36
|
Lung underdevelopment is a rare congenital anomaly with variable clinical significance and presenting symptoms. It usually manifests during childhood. We present two cases of developmental lung anomaly subtypes and discuss clinical presentation and outcomes in such patient populations.
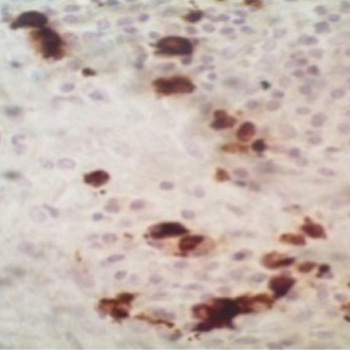
|
Views: 43
HTML: 11
PDF: 36
|
We report a rare yet successful utilisation of anti-CD20 therapy using rituximab for treatment of a case of IgG4-related mastitis proven by clinical, serological, and histopathological evidence. This was affecting a mid-aged female who was referred to the rheumatology clinic by the breast surgeons to help assessing for the possibility of an underlying inflammatory process involving the breast tissue unilaterally.
The clinical course was apparently complex with an onset of an induration in the right lateral superior quadrant of the breast with mild discomfort and heaviness sensation. This increased over a course of 2 weeks before presentation to the general surgery clinic.
Subsequent investigations confirmed that the case was IgG4-related mastitis and a trial of steroids and disease modifying anti-rheumatic drugs (DMARDs) was partially helpful, but not to a full degree, mandating the utilisation of a more advanced mode of therapy, so rituximab was selected.
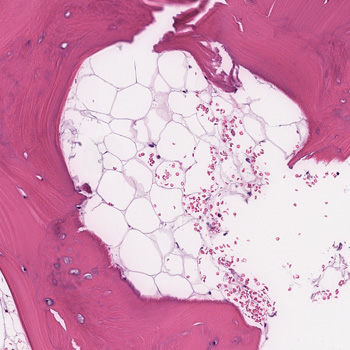
|
Views: 128
HTML: 14
PDF: 71
|
Background: Autoimmune diseases are not contraindications for immune checkpoint inhibitors (ICI) therapy in patients with cancer. However, immune-related adverse events (irAEs) are frequently observed in patients receiving ICIs including dermatitis, thyroiditis, colitis, and pneumonitis. Thrombocytopenic purpura, aplasia, and haemophagocytic lymphohistiocytosis (HLH) are rarely observed during ICIs.
Case description: We report the case of a male patient with pre-existing untreated HLA B27 and ankylosing spondylitis with gastric cancer and liver metastases. The 79-year-old man was treated with anti-HER2 trastuzumab and anti-PD-1 nivolumab. Seventeen days after the seventh cycle of treatment, he presented at the emergency department with acute fever, confusion, and hypotension. Laboratory results showed pancytopenia, and elevation of ferritin and triglyceride. No infections were detected. Although not seen in a bone marrow biopsy, clinical presentation, and absence of infection, together with an H-score of 263, indicated HLH. The patient was treated with dexamethasone for four days and discharged on a tapering dose of steroids. At the two-month follow-up, clinical presentation was normal and blood test almost normalised. At 8 months, no liver metastases were observed.
Conclusions: In a patient with a pre-existing autoimmune condition, immunotherapy led to the development of HLH, which was controlled by glucocorticoid. Absence of the feature of haemophagocytosis in the bone marrow biopsy did not exclude the diagnosis, as HLH can occur in the spleen or in the liver. Glucocorticoid therapy did not prevent the anti-cancer effect of ICIs, and liver metastases disappeared 8 months post-HLH. This case warrants further research on the interplay between autoimmunity and ICI response, as well as ICI-induced irAEs.
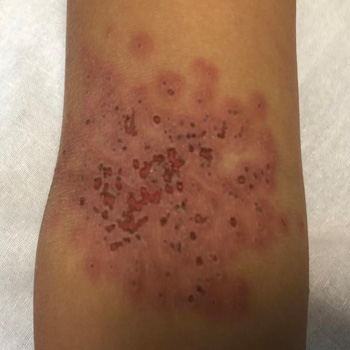
|
Views: 176
HTML: 16
PDF: 90
|
Introduction: Kaposi’s varicelliform eruption (KVE), also known as eczema herpeticum or eczema vaccinatum, is an acute dermatosis that affects patients with chronic dermatopathies. The diagnosis is primarily clinical and is characterised by the presence of a vesicular exanthema on physical examination. The exanthema subsequently evolves into crusted lesions with typical circular ulcerations in ‘punched-out’ areas on the skin affected by the underlying dermatopathy.
Case description: We present the case of a 6-year-old patient who presented to the Paediatric Emergency department with skin lesions consistent with eczema herpeticum. The patient’s management was initially outpatient; however, due to the slow progression of the condition, hospitalisation and intravenous antiviral treatment were initiated.
Discussion: KVE affects patients with chronic dermatoses, especially atopic dermatitis. It is important to know the clinical presentation for an early suspicion. KVE is a medical emergency that requires prompt diagnosis and treatment. It can progress to secondary viraemia, which can be fatal in up to 10% of immunocompetent individuals and up to 50% of immunocompromised individuals. It is important to be aware of this condition and to start early treatment with antivirals, especially given the high prevalence of atopic dermatitis in our population. This condition is one of the most serious complications that can occur in these patients.
| 2.1 = | 1.730 Cit. to date |
| 842 Docs. to date |




Publisher
Official Journal of the
European Federation of Internal Medicine
www.efim.org
Publisher: SMC media Srl
Via Giovenale, 7 - 20136 Milan - Italy
P.IVA 07626490960
info@ejcrim.com
www.ejcrim.com - ISSN: 2284-2594 - © EFIM 2014-2023, Published by SMC Media srl, Italy - Privacy policy

