EJCRIM 2023 CiteScore
| 2.1 = | 1.762 Cit. to date |
| 842 Docs. to date |
Last updated on 05 May, 2024
Updated monthly
Updated monthly
Powered by


|
Views: 493
HTML: 62
PDF: 224
|
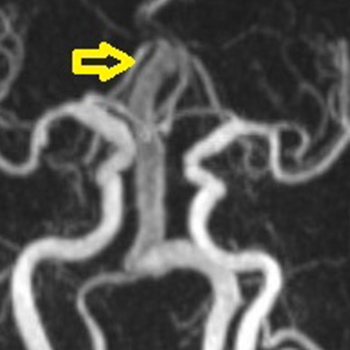
Vertebro-basilar dolichoectasia (VBD) is a rare pathology of unknown aetiology. Its clinical presentation is wide and prognosis is generally poor with a high mortality rate. Cerebral magnetic resonance imaging is the gold standard for diagnosis.
We report an unusual case of intracranial dolichoectasia. VBD was revealed during investigation of a patient with altered mental status. CT brain imaging demonstrated severe obstructive hydrocephalus secondary to compression of the third ventricle. Management is always challenging and depends on the location and the mode of presentation. Our patient died despite surgical management with placement of an external ventricular shunt
|
Views: 591
HTML: 105
PDF: 349
|
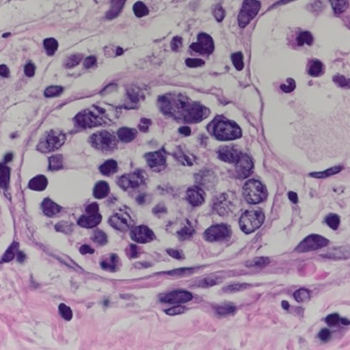
Introduction: Delayed diagnosis of intravascular large B-cell lymphoma (IVLBCL) is associated with a poor prognosis, making early diagnosis and treatment essential. However, early diagnosis remains challenging.
Case description: Here we present the case of a 75-year-old man with fever of unknown origin, in whom random skin biopsy (RSB) allowed early diagnosis of IVLBCL.
Discussion: The usefulness of RSB, which involves incisional skin biopsies of three or more sites that contain subcutaneous fatty tissue, such as the thighs, abdomen and upper arms, has been debated. In cases of suspected IVLBCL, RSB is less invasive than a biopsy of the internal organs.
Conclusion: We suggest that combining RSB with bone marrow examination may facilitate the diagnosis of IVLBCL.
|
Views: 554
HTML: 643
PDF: 363
|
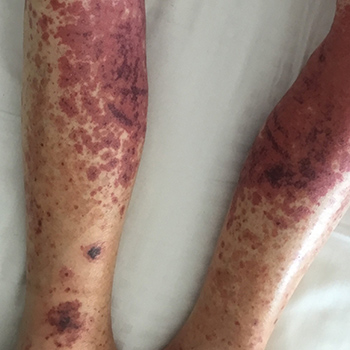
Leukocytoclastic vasculitis is a cutaneous, small-vessel vasculitis. In 50% of cases the aetiology is idiopathic but it can be linked to drugs, infections, autoimmune disorders and various types of cancer. Levamisole is used as an adulterant in cocaine and heroin and has been associated with the development of leukocytoclastic vasculitis. We describe an atypical presentation of a patient with levamisole-induced leukocytoclastic vasculitis who presented with diffuse skin abscesses and a purpuric rash of the upper and lower limbs.
|
Views: 740
HTML: 149
PDF: 428
|
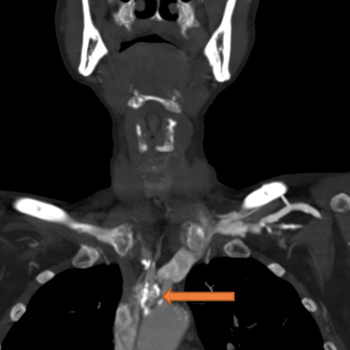
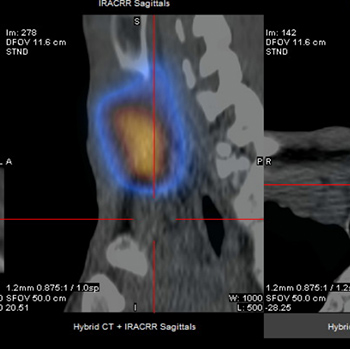
Introduction: Subclavian artery stenosis (SAS) is a manifestation of peripheral artery disease (PAD). Presentation varies, ranging from arm claudication and muscle fatigue to symptoms which reflect vertebrobasilar hypoperfusion, among which are syncope, ataxia and dysphagia. Although rare, severe bilateral SAS can exist and present as refractory hypotension. We describe a case of bilateral SAS masquerading as circulatory shock, or rather ‘pseudoshock’.
Case Description: A 59-year-old female patient presented to the emergency department with complaints of dark stools. She was anaemic and hypotensive and therefore suspected to have an acute gastrointestinal bleed (GIB) with resultant haemorrhagic shock. Her hypotension was unresponsive to fluid resuscitation and blood transfusions. Bilateral upper extremity radial artery catheters confirmed low blood pressures. After her blood pressure failed to improve despite the addition of several vasopressors, a femoral artery catheter (FAC) was placed, which revealed significant hypertension discordant with the hypotension measured by the radial artery catheters. Review of CT angiography of the upper extremities revealed the presence of bilateral SAS which was deemed to be the aetiology of the falsely low blood pressure.
Discussion: SAS should be suspected in patients with lower extremity PAD or a blood pressure (BP) differential of 15 mmHg or more between arms. When bilateral subclavian arteries are stenosed, this difference in BP may be concealed, making lower extremity BP measurements, as seen in non-invasive tests such as ankle brachial index (ABI) tests or through more invasive procedures such as FAC placement, critically important.
Conclusion: Bilateral SAS may present as pseudo-hypotension. In cases of refractory shock of unclear aetiology, especially in patients with known PAD, a high index of suspicion is warranted for ‘pseudoshock’ secondary to severe vascular stenosis. Comparison of upper and lower extremity BP via invasive arterial catheters or non-invasive ABI tests can aid in the diagnosis of bilateral SAS.
|
Views: 590
HTML: 56
PDF: 582
|
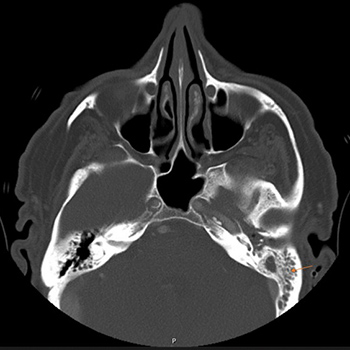
Graves’ disease is an autoimmune disorder that results in hyperthyroidism, caused by autoantibodies to the thyrotropin receptor (TRAbs) stimulating thyroid hormone synthesis, giving rise to a variety of systemic manifestations such as goitre, dermatopathy and orbitopathy.
The authors present the case of a 28-year-old man admitted to hospital for a 3-week history of fatigue, shortness of breath, palpitations and diffuse goitre, after recent mild SARS-CoV-2 infection. Laboratory investigation revealed hyperthyroidism with TRAbs elevation. Thyroid ultrasound confirmed a diffusely heterogeneous and irregular thyroid gland and a nodular image below the sternal notch. Thyroid scintigraphy excluded the nodule and confirmed a Graves’ disease pattern. Following the initiation of methimazole, the patient had complete resolution of symptoms and normalization of thyroid values.
The results suggest a possible association between Graves’ disease and SARS-CoV-2 infection acting as a trigger. Graves’ disease is an important differential diagnosis to keep in mind when patients present with hyperthyroidism after COVID-19 disease.
|
Views: 383
HTML: 188
PDF: 172
|
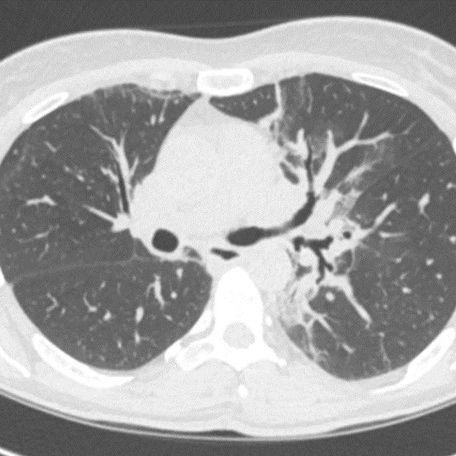
Gradenigo syndrome (GS) was described primarily in the paediatric population, especially in the pre-antibiotic era. GS is rarely reported in the elderly population, especially in the post-antibiotic era. We present the rare case of a 67-year-old man who presented with an incomplete triad of symptoms (without abducens nerve palsy) of GS that failed medical therapy and was successfully treated with surgical intervention (mastoidectomy and petrous apicectomy). Physicians should be familiar with atypical presenting symptoms of GS as it can lead to life-threatening complications, especially in the elderly. GS cases resistant to medical therapy may require prompt appropriate imaging studies and surgical intervention.
|
Views: 853
HTML: 150
PDF: 387
|
A patient with antisynthetase syndrome with pulmonary and muscular involvement was treated with immunosuppressive agents without corticosteroids. Rituximab was added to mycophenolate mofetil therapy with improvement in lung functional and imaging findings and normalization of creatine kinase levels.

|
Views: 417
HTML: 105
PDF: 257
|
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) is characterised by skin rash together with visceral organ involvement, lymphadenopathy, eosinophilia and atypical lymphocytosis. The syndrome is clinically heterogeneous, making diagnosis challenging. It has an annual incidence of 2 per 100,000 population and a mortality rate of 2–10%. We describe the first case of DRESS induced by certolizumab, a biologic disease-modifying antirheumatic drug (bioDMARD).

|
Views: 754
HTML: 334
PDF: 513
|
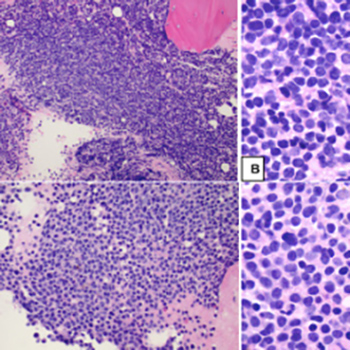
The mRNA-1273 SARS-CoV-2 vaccine received emergency use authorization in December 2021. We present a case of myasthenia gravis (MG) which became clinically apparent following vaccination against SARS-CoV-2. A 30-year-old man developed acute onset diplopia, 2 days after receiving his first mRNA-1273 vaccination against SARS-CoV-2. He reported blurred vision with horizontally displaced images, which worsened with increased eye strain. Diplopia resolved when one eye was covered. He also had fatigable arm weakness, but denied dysphagia, dysarthria, dysphonia or dyspnoea. On examination, he had left-sided ptosis and esotropia at rest which worsened with sustained upward gaze and prolonged focus. He also had fatigable weakness of neck flexion and extension (4+/5), and generalized, fatigable weakness (4/5). His single-breath count was 38. Cranial nerves, sensory examination and deep tendon reflexes were normal. A 2-min ice-pack test and neostigmine test temporarily improved his diplopia and ptosis. The acetylcholine receptor (AChR) antibody was borderline high and muscle-specific tyrosine kinase (MuSK) antibody was negative. Chest CT and brain MRI with contrast were unremarkable. The patient was diagnosed with MG and oral pyridostigmine and prednisone therapy were initiated.
We present a case of newly diagnosed MG after administration of mRNA-1273 vaccination against SARS-CoV-2. Although there has been long-standing discussion regarding the potential for vaccines to exacerbate autoimmune conditions, data remain sparse and consensus has not been reached. Consequently, this case is important to make providers aware of potential side effects of a novel vaccine, and may also help guide the selection of vaccination candidates and monitoring parameters.
|
Views: 549
HTML: 80
PDF: 348
|
It is rare for IgM multiple myeloma (MM) and mantle cell lymphoma (MCL) to coexist. Furthermore, it is challenging to demonstrate if there are two distinct types of neoplasia or if plasma cell differentiation of MCL is present. We discuss the case of a patient concomitantly diagnosed with MCL and IgM MM, and the subsequent diagnostic and management difficulties, and the positive treatment outcome.
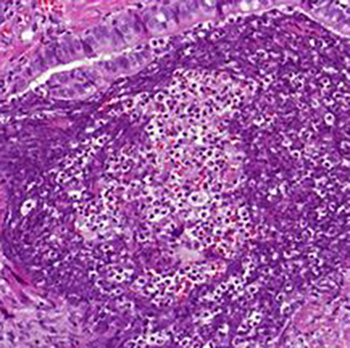
|
Views: 534
HTML: 84
PDF: 276
|
Background: Chronic granulomatous disease (CGD) is a rare immunodeficiency disorder resulting in phagocytic cell dysfunction. It is characterized by deficient cellular immunity against bacteria and fungi, and an excessive inflammatory response resulting in granuloma formation. It manifests, usually in early childhood, with recurrent bacterial and fungal infections or inflammatory complications. The infections, such as invasive pulmonary aspergillosis, can be life-threatening.
Case description: Our patient was a 40-year-old man with no pulmonary history who presented with bilateral pulmonary nodules and pronounced eosinophilia in peripheral blood and bronchoalveolar lavage fluid, mimicking eosinophilic pneumonia. During treatment with corticosteroids, the patient deteriorated clinically and radiographically. Extensive investigations failed to provide a diagnosis. A lung biopsy demonstrated the presence of granulomas and Aspergillus fumigatus hyphae. Advanced screening to detect underlying immunodeficiency revealed CGD.
Discussion: This case report describes a unique first presentation of CGD. It reminds physicians of the possibility of CGD as an underlying immune disorder in invasive aspergillosis and highlights the challenges of diagnosing invasive pulmonary aspergillosis. We discuss the diagnostic pitfalls of this case and propose a diagnostic work-up for eosinophilic lung disease.

|
Views: 1287
HTML: 107
PDF: 287
|
A 23-year-old Japanese woman presented with a 1-month history of dyspnoea and chest discomfort. Since the symptoms improved with dynamic and sensory stimulation and also caused insomnia, we considered a variant of restless legs syndrome (RLS) called ‘restless chest syndrome’, although there were no symptoms in the extremities. We initiated oral administration of pramipexole 0.25 mg daily, and her symptoms, including dyspnoea, chest discomfort and insomnia, improved within 1 week. RLS should be considered in the differential diagnosis in patients who present with abnormal sensations that worsen at night, with insomnia, regardless of the site of the symptoms.

|
Views: 486
HTML: 55
PDF: 265
|
Introduction: Severe haemophilia A is characterized by serious factor VIII deficiency (biological activity <1%) resulting in frequent spontaneous haemorrhage and abnormal bleeding after minor injury, surgery or tooth extraction.
Patient and Methods: We report the case of a 58-year-old patient with severe haemophilia A without inhibitors but with other comorbidities (HCV and HIV seroconversion), who underwent coronary angioplasty and stent implantation after acute myocardial infarction.
Results: Compared with previous therapy, Nuwiq® led to a reduction of about 20% in drug consumption (360,000 IU vs 540,000 IU per year) and in the annualized bleeding rate (ABR) (5 vs 15).
Discussion: Pharmacokinetic-guided personalized prophylaxis with Nuwiq® provided bleeding protection with good tolerability and a satisfactory pharmacokinetic profile in a patient with severe haemophilia A and comorbidities whose replacement therapy had to be adjusted because of other contraindicated treatment.
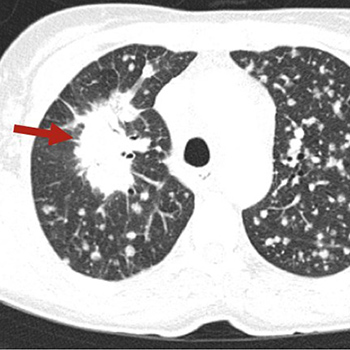
|
Views: 603
HTML: 66
PDF: 278
|
Arterial thrombosis and Budd–Chiari syndrome are rare conditions in lung cancer patients. We report the case of a 53-year-old woman who presented with respiratory symptoms, lumbar pain, weight and appetite loss, and an x-ray showing a lung nodule and diffuse micro-opacities. She was diagnosed with lung neoplasia with extensive lung, liver, lymph node and bone metastases. After discharge she was readmitted with a respiratory infection, and as her condition deteriorated, computed tomography was performed and revealed ischaemic areas in the spleen and kidney, and venous thrombosis, related to Budd–Chiari syndrome, with hepatic ischaemia. Despite hypocoagulation, her clinical condition deteriorated and she died soon afterwards.
| 2.1 = | 1.762 Cit. to date |
| 842 Docs. to date |




Publisher
Official Journal of the
European Federation of Internal Medicine
www.efim.org
Publisher: SMC media Srl
Via Giovenale, 7 - 20136 Milan - Italy
P.IVA 07626490960
info@ejcrim.com
www.ejcrim.com - ISSN: 2284-2594 - © EFIM 2014-2024, Published by SMC Media srl, Italy - Privacy policy

