EJCRIM 2023 CiteScore
| 2.1 = | 1.762 Cit. to date |
| 842 Docs. to date |
Last updated on 05 May, 2024
Updated monthly
Updated monthly
Powered by


|
Views: 718
HTML: 45
PDF: 431
|
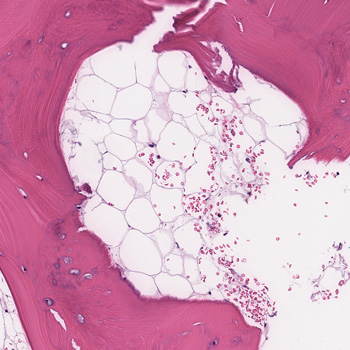
Background: Autoimmune diseases are not contraindications for immune checkpoint inhibitors (ICI) therapy in patients with cancer. However, immune-related adverse events (irAEs) are frequently observed in patients receiving ICIs including dermatitis, thyroiditis, colitis, and pneumonitis. Thrombocytopenic purpura, aplasia, and haemophagocytic lymphohistiocytosis (HLH) are rarely observed during ICIs.
Case description: We report the case of a male patient with pre-existing untreated HLA B27 and ankylosing spondylitis with gastric cancer and liver metastases. The 79-year-old man was treated with anti-HER2 trastuzumab and anti-PD-1 nivolumab. Seventeen days after the seventh cycle of treatment, he presented at the emergency department with acute fever, confusion, and hypotension. Laboratory results showed pancytopenia, and elevation of ferritin and triglyceride. No infections were detected. Although not seen in a bone marrow biopsy, clinical presentation, and absence of infection, together with an H-score of 263, indicated HLH. The patient was treated with dexamethasone for four days and discharged on a tapering dose of steroids. At the two-month follow-up, clinical presentation was normal and blood test almost normalised. At 8 months, no liver metastases were observed.
Conclusions: In a patient with a pre-existing autoimmune condition, immunotherapy led to the development of HLH, which was controlled by glucocorticoid. Absence of the feature of haemophagocytosis in the bone marrow biopsy did not exclude the diagnosis, as HLH can occur in the spleen or in the liver. Glucocorticoid therapy did not prevent the anti-cancer effect of ICIs, and liver metastases disappeared 8 months post-HLH. This case warrants further research on the interplay between autoimmunity and ICI response, as well as ICI-induced irAEs.
|
Views: 185
HTML: 9
PDF: 153
|
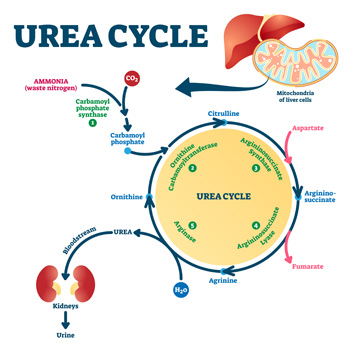
Background: Hyperargininemia is a rare inherited metabolic disorder of the urea cycle with an autosomal recessive transmission. It occurs due to a deficiency of the enzyme arginase I and causes progressive neurological damage. Very few cases are diagnosed in adulthood, with the majority being diagnosed before the age of 4. Currently, this condition is diagnosed by a mass spectrometry technique in neonatal screening, which has been implemented in Portugal since 2007; births before that were not screened for this entity.
Case description: We present a case of a 23-year-old woman referred to the internal medicine and neurology departments with a history of two hospital admissions for rhabdomyolysis at the age of 18, consanguineous parents, learning difficulties and multiple falls since the age of 8. In addition, the patient also had behavioural changes so she had psychological counselling at school, but lacked family support. Neurological examination showed mild proximal paraparesis, and spastic and paraparetic gait. The aetiological study revealed a pathological variant in homozygosity ARG1 and increased blood levels of arginine. Therefore, the diagnosis of hyperargininemia was confirmed.
Conclusions: Compared to other urea cycle disorders, hyperargininemia is the rarest one. It is important to recognise the characteristic clinical features and diagnose it early because a favourable outcome can be achieved with appropriate treatment. This case shows a delayed diagnosis of hyperargininemia and highlights the importance of the internist’s role in diagnosing rare diseases.
|
Views: 155
HTML: 25
PDF: 128
|
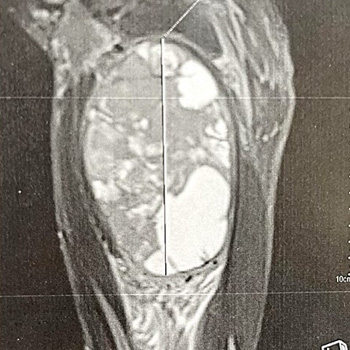
Background: Patients with neurofibromatosis type I (NF1) have an increased risk of developing soft-tissue sarcomas, particularly those related to the nervous system. Epithelioid sarcoma (ES) is an exceptionally rare subtype of soft-tissue sarcoma, with limited knowledge about its clinical presentation and optimal management in NF1. This report aims to provide insights into the characteristics and outcomes of ES in NF1 patients.
Case description: A 37-year-old man with a history of NF1 presented with a progressively worsening mass on his right inner thigh. An MRI scan revealed a well-defined tissue mass originating from the adductor magnus muscle, later confirmed as ES through histopathology and immunohistochemistry. Considering poor local and general prognosis, the multidisciplinary team recommended salvage hip disarticulation, however the patient refused and opted for palliative marginal resection to reduce the tumour size. The patient’s condition declined rapidly, and he succumbed six days after the surgery.
Conclusion: This case highlights the rarity of ES in NF1 patients and underscores the potential for malignant tumour development in this population. Further research is needed to improve our understanding and management of sarcomas in the context of NF1.
|
Views: 266
HTML: 52
PDF: 231
|
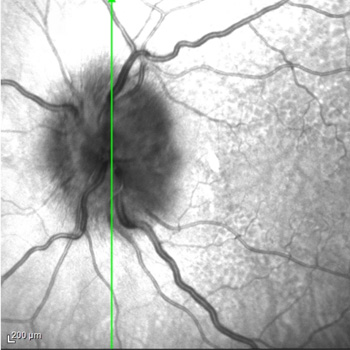
Background: eosinophilic granulomatosis with polyangiitis (EGPA) is a rare multisystem inflammatory disease characterized by asthma, eosinophilia and granulomatous or vasculitic involvement of various organs. While the eye is uncommonly affected in patients with EGPA, multiple ophthalmic manifestations have been reported, which can result in serious visual impairment without timely treatment.
Case report: we report the case of a 79-year-old woman with a history of asthma and nasal polyps who presented with low-grade fever, mild alteration of mental status, and fatigue. Chest X-ray revealed bilateral interstitial infiltrates. Lab tests showed elevated C-reactive protein level and eosinophilia (eosinophil count, 4.6 x109 cells/l); blood cultures and parasitological examination of stools tested negative. Four days after presentation, the patient reported sudden and severe blurring of vision in her left eye. Ophthalmological examination revealed bilateral swollen optic disc and visual field loss, more severe in the left eye. A diagnosis of EGPA complicated by arteritic anterior ischaemic optic neuropathy (A-AION) was proposed, while an alternative or concurrent diagnosis of giant cell arteritis was ruled out based on clinical picture.
Immunosuppressive treatment with high-dose intravenous glucocorticoids was promptly started. The patient’s visual defect did not improve; however, two months later, no worsening was registered on ophthalmic reassessment.
Conclusions: A-AION is an infrequent but severe manifestation of EGPA, requiring prompt diagnosis and emergency-level glucocorticoid therapy to prevent any further vision loss. Disease awareness and a multidisciplinary approach are crucial to expedite diagnostic work-up and effective management of EGPA-related ocular complications.
|
Views: 210
HTML: 15
PDF: 200
|
Introduction: Most pregnancies in women after a kidney transplant result in a live birth, but kidney functions should be stable for one year before conception. For immunosuppression modification occurring before pregnancy, azathioprine is used because it is considered safe for major congenital malformations during pregnancy. However, there may be an association between exposure to azathioprine during pregnancy and the onset of an unusual, early and severe form of intrahepatic cholestasis.
Case description: A young patient with a twin pregnancy after a second kidney transplant experienced intrahepatic cholestasis. There was a wide range of differential diagnosis. A battery of tests was requested including autoimmune markers, virology, and imaging. The conclusion that azathioprine was contributing to intrahepatic cholestasis with pregnancy was reached after exclusion of all other differentials.
Conclusions: Complications of pregnancy after a kidney transplant include hypertension, pre-eclampsia, deterioration of graft function up to rejection, but also unusual side effects of immunosuppression medication.
|
A case of amyloid goitre in heavy chain amyloidosis: diagnostic challenges and clinical implications
Views: 202
HTML: 26
PDF: 172
|
Immunoglobulin heavy chain amyloidosis (AH amyloidosis) is an extremely rare subtype of immunoglobulin-derived amyloidosis and there is limited literature on how to diagnose and manage this disorder. We describe a rare case of AH amyloidosis with amyloid goitre and the importance of mass spectrometry in the identification of the different types of amyloids. While additional studies are needed, several observations suggest important practical implications, including differences in clinical picture, prognosis, and pathologic diagnosis.

|
Views: 288
HTML: 52
PDF: 187
|
Chronic lymphocytic leukaemia (CLL) is a lymphoproliferative disorder characterised by an accumulation of monoclonal B lymphocytes, with an increased risk of secondary cancers. The coexistence of CLL and chronic myeloid leukaemia (CML) is a rare phenomenon, with three main types being classified: CML preceding CLL, CLL preceding CML and simultaneous occurrence. The coexistence of these chronic leukaemias poses a complex clinical challenge, with the underlying mechanisms of their association remaining enigmatic. Here, we present a report of an elderly male with a long history of CLL, who was subsequently diagnosed with secondary CML.
|
Views: 265
HTML: 14
PDF: 217
|
Castleman’s disease (CD) and thrombotic thrombocytopenic purpura (TTP) are rare diseases that can affect the general population, especially those with HIV. Owing to their rarity, the association between CD and TTP remains insufficiently understood. In this study, we present a case of a 53-year-old patient with controlled HIV infection who presented with fever, lymphadenopathy, severe anaemia, and thrombocytopenia. After a series of tests, the diagnosis was concurrent human herpesvirus 8 (HHV8)-related multicentric CD (MCD) and TTP. Only four male patients were previously reported having this association, with HHV8 present in four and HIV in three patients, suggesting that coinfection with HHV8 and HIV is a pivotal factor in MCD with TTP occurrence.
|
Views: 194
HTML: 9
PDF: 189
|
Spontaneous bleeding into the upper airways is a rare and potentially life-threatening complication of chronic anticoagulation. There are scarce cases in the literature demonstrating upper airway haematomas secondary to warfarin use, which is the predominant anticoagulant used by clinicians despite having a complex pharmacokinetic and pharmacodynamic profile. We report a compelling case featuring warfarin-induced sublingual haematoma, managed conservatively through the reversal of anticoagulation using fresh frozen plasma complemented by vigilant monitoring within the Intensive Care Unit (ICU).
|
Views: 290
HTML: 46
PDF: 266
|
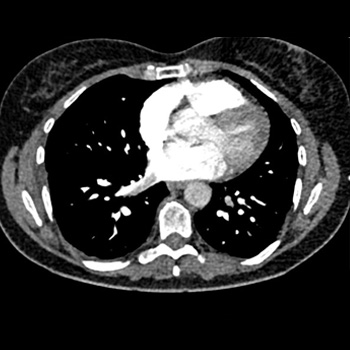
A 52-year-old female with a history of chronic lymphoedema and untreated deep vein thrombosis, presented with non-specific right-sided chest pain. A CT angiogram confirmed bilateral inferior pulmonary vein thromboses (PVT). A comprehensive hypercoagulable workup and age-appropriate cancer screening were unremarkable; the lack of associated risk factors confirmed idiopathic PVT. The management strategy of systemic anticoagulation with apixaban and multidisciplinary follow-up underscores the treatment challenges of rare presentations. This case accentuates the importance of considering PVT in differential diagnoses of atypical chest pain and contributes valuable insights into the diagnosis, understanding and management of this uncommon condition.
| 2.1 = | 1.762 Cit. to date |
| 842 Docs. to date |




Publisher
Official Journal of the
European Federation of Internal Medicine
www.efim.org
Publisher: SMC media Srl
Via Giovenale, 7 - 20136 Milan - Italy
P.IVA 07626490960
info@ejcrim.com
www.ejcrim.com - ISSN: 2284-2594 - © EFIM 2014-2024, Published by SMC Media srl, Italy - Privacy policy

