EJCRIM 2023 CiteScore
| 2.1 = | 1.762 Cit. to date |
| 842 Docs. to date |
Last updated on 05 May, 2024
Updated monthly
Updated monthly
Powered by



|
Views: 0
HTML: 0
PDF: 0
|
Tumour-to-tumour metastasis (TTM) is a rare phenomenon that clinicians should be aware of when evaluating patients with a history of prostate cancer. We present the diagnosis and management of an 80-year-old former smoker with high-risk prostate cancer, who developed a lung nodule consistent with TTM. The patient had concurrent primary lung adenocarcinoma and metastatic prostate cancer, making this a unique case of dual primary and metastatic malignancies. The complexity of this case highlights the need for comprehensive evaluation and interdisciplinary management in patients with multiple malignancies. The literature review reveals that these are extremely rare occurrences, with most cases involving metastasis to the second primary tumour. Despite the challenges in diagnosing preoperatively, it is important to consider TTM as a possibility in patients with prostate cancer who present with a lung nodule. This report presents one of the few documented cases of TTM. It also reviews relevant cases in the literature and discusses the current situation in relation to established criteria for classifying combination tumours.

|
Views: 0
HTML: 0
PDF: 0
|
Haemorrhagic pleural effusion can be a challenging diagnosis that requires a thorough investigation and sometimes a multidisciplinary team of physicians to reach the underlying aetiology. Causes can include pulmonary malignancy, pulmonary infections, connective tissue diseases, asbestos associated, intra-abdominal conditions such as pancreatitis and ovarian tumours, cardiovascular disorders such as ruptured aneurysms and pulmonary infarction, as well as other miscellaneous causes. One such cause is endometriosis in the thoracic cavity. Endometriosis is a chronic illness associated with the occurrence of endometrial tissue outside the endometrium. Insertion of endometrial tissue in the thoracic cavity is rare, with only a few cases described. This case report gives detail of a 30-year-old nulligravida suspected of having thoracic endometriosis following a history of catamenial dyspnoea and associated pleural effusion. The diagnosis was confirmed through the histopathological study of tissue obtained via thoracoscopic surgery. Excision of the endometrial tissue was done, and the patient then continued medical treatment with progestins and gonadotrophin-releasing hormone (GnRH) agonists. Following therapy, the index patient was asymptomatic. A multidisciplinary approach is often needed in the diagnosis and management of thoracic endometriosis, involving both medical and surgical specialities. Minimally invasive surgery is the gold standard of diagnosis, allowing for direct visualisation of implants and nodules and should be followed by medical treatment to reduce the risk of recurrence. Medical therapy alone is associated with higher rates of recurrence. Physicians must have a high degree of suspicion as thoracic endometriosis is a disease that can often be missed.

|
Views: 70
HTML: 13
PDF: 36
|
Pneumocystis jirovecii is an opportunistic fungus that infects the lungs but can involve other organs, including the skin and lymph nodes. Risk factors include human immunodeficiency virus (HIV), solid organ/haematological malignancies and a CD4 cell count of fewer than 200 cells/µl. Pneumocystis jirovecii pneumonia (PJP) infection is reported less frequently these days with the advent of prophylaxis with trimethoprim-sulfamethoxazole (TMP-SMX).
We report a case of extrapulmonary PJP infection in a patient while receiving pentamidine prophylaxis in a T-cell prolymphocytic leukaemia, who underwent an allogeneic stem cell transplant. There are plenty of reported cases of PJP on pentamidine prophylaxis; however, none had cutaneous PJP infection. Cutaneous P. jirovecii infection (CPJ) is an extrapulmonary infection that is rarely reported. Our patient’s skin biopsy was inconclusive, but the skin nodules improved once he was initiated on TMP-SMX. Many transplant patients cannot tolerate TMP-SMX for various reasons and are placed on second-line prophylaxis for PJP, which does not prevent extrapulmonary PJP infections. Our case highlights the challenges of diagnosing such a rare infection in immunocompromised patients. Extrapulmonary PJP should be suspected in patients with a history of pulmonary PJP and persistent elevated Fungitell® levels in low CD4 counts.

|
Views: 235
HTML: 10
PDF: 126
|
Background: Alagille syndrome (ALGS) is a multisystem disorder involving at least three systems among the liver, heart, skeleton, face, and eyes. Common cardiac associations include pulmonary artery stenosis/atresia, atrial septal defect (ASD), ventricular septal defect (VSD) and tetralogy of fallot (ToF). Coarctation of aorta (CoA), renal and intracranial arteries are commonly involved vessels in Alagille syndrome. We present two cases with rare cardiovascular manifestations of Alagille syndrome.
Case description: Case 1: A 25-year-old female with a history of Alagille syndrome presented to the cardiologist office for progressive exertional dyspnoea, orthopnoea, and palpitations. She was tachycardiac on examination and had an apical diastolic rumble. A transthoracic echocardiogram (TTE) showed a left ventricular ejection fraction (LVEF) of 60% and parachute mitral valve (PMV) with severe mitral stenosis. A transoesophageal echocardiogram (TOE) showed insertion of chordae into the anterolateral papillary muscle, severe mitral stenosis with a valve area of 0.7 cm. She was referred to a congenital heart disease specialist and underwent robotic mitral valve replacement with improvement in her symptoms.
Case 2: A 27-year-old female with known Alagille syndrome and resistant hypertension presented to the cardiologist office due to progressive exertional dyspnoea for a year. She was hypertensive and had a new 2/6 systolic ejection murmur along the left upper sternal border. TTE revealed an LVEF of 60% and pulmonary artery pressure of 19 mmHg. A CoA was suspected distal to the left subclavian artery due to a peak gradient of 38 mmHg. Cardiac magnetic resonance (CMR) imaging ruled out CoA, and diffuse narrowing of the descending thoracic aorta measuring 13–14 mm in diameter was noted. The patient was referred to a congenital heart disease specialist for further management.
Conclusion: PMV presenting as mitral stenosis and mid-aortic syndrome are not commonly described anomalies in association with Alagille syndrome. TTE, TOE and CMR played a key role in diagnosis and management of these patients.

|
Views: 288
HTML: 14
PDF: 140
|
Background: Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease, characterised by multi-organ affections. Haematological involvement is a common manifestation of SLE, consisting of autoimmune peripheral cytopenia. Autoimmune myelofibrosis (AIMF) is a rare cause of cytopenia in SLE; it could precede or be concurrent with the diagnosis of SLE. There are few studies that describe this association.
Case description: We report a case of AIMF revealing the diagnosis of SLE in a 34-year-old female, presented with episodes of gingival bleeding associated with peripheral inflammatory polyarthralgia, photosensitivity and deterioration of general condition. Clinical examination revealed a soft pitting oedema in the lower limbs. Laboratory investigations showed a pancytopenia, inflammatory biological syndrome, with positive 24-hour proteinuria and anti-native DNA antibodies. A bone marrow biopsy showed diffuse myelofibrosis associated with maturation disorders and no tumour infiltrate. Renal biopsy revealed proliferative glomerulonephritis class III with immune deposits.
Conclusion: The association of AIMF with SLE has been rarely reported, and it could be another cause for cytopenia in SLE.

|
Views: 261
HTML: 14
PDF: 167
|
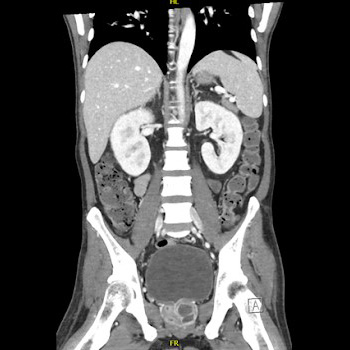
Introduction: Blue rubber bleb nevus syndrome is a rare disorder of venous malformations, with around 200 cases reported. We present a case of Mycobacterium xenopi infection in a patient with blue rubber bleb nevus syndrome.
Case Description: A 40-year-old female with blue rubber bleb nevus syndrome, asthma, and bronchiectasis came to the pulmonology clinic with shortness of breath and a cough. She was recently admitted for a bronchiectasis exacerbation but continued to have a worsening productive cough and fevers. The most recent CT scan of the chest showed interval stable right upper lobe fibrocavitary disease, demonstrating gradual progression over two years. She had occasional positive cultures for Mycobacterium Avium Complex and M. xenopi one year previously, assumed to be a colonizer and not treated. Most recent hospital cultures were negative for bacteria and an acid-fast bacilli smear. She was sent to the emergency department for bronchiectasis exacerbation and returned to the clinic six weeks later with two sputum cultures growing M. xenopi. It was decided to treat M. xenopi as this was likely the cause of her cavitary lung lesion and frequent infections. Azithromycin, rifampin, and sulfamethoxazole/trimethoprim were initiated. Intravenous amikacin was added later on. She finally had a right partial lung resection done after one year at an outside hospital. She was on and off antibiotics for M. xenopi for approximately three years with negative repeat cultures for non-tuberculous mycobacteria.
Conclusion: Due to the high mortality of M. xenopi infections (which can be as high as 69%), treatment of at least twelve months is recommended. To our knowledge, this is the first reported case of M. xenopi in a patient with blue rubber bleb nevus syndrome.
|
Views: 309
HTML: 40
PDF: 189
|
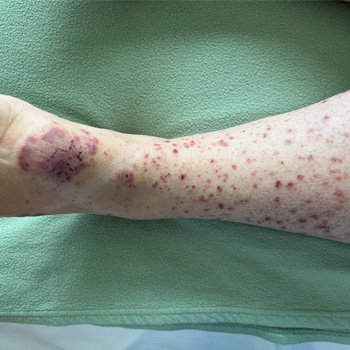
Background: Melioidosis is an infection caused by Burkholderia pseudomallei, a Gram-negative bacterium. It is a disease endemic to Southeast Asia and northern Australia although its global incidence has been rising. It most commonly infects people with certain identified risk factors such as diabetes, alcoholism, thalassemia, and underlying chronic disease involving lungs, kidney and liver. This bacterium is capable of producing a wide array of clinical manifestations ranging from asymptomatic disease to localised infections such as in the lung, bone or skin to disseminated infection.
Case description: This is a case, from United Arab Emirates, of a 40-year-old male recently diagnosed with diabetes who presented with multiple abscesses and was eventually diagnosed with disseminated melioidosis. He was treated successfully with antibiotics and drainage of abscesses.
Conclusion: In non-endemic regions, melioidosis can be easily missed in common diagnostic approaches. This gap of awareness could delay the diagnosis and allow further deterioration of the patient due to complications. Thus, case reports like this can enlighten internists about changing incidences and complexity of clinical presentations, thus preparing them to better handle such patients in the future.
|
Views: 153
HTML: 11
PDF: 153
|
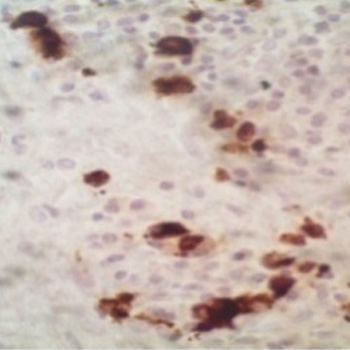
Background: Annular erythema is a rare manifestation of leukocytoclastic vasculitis. It may be associated with various drugs, infections, malignancies, or systemic diseases.
Case description: A 36-year-old woman with no personal medical history presented with annular erythema with target lesions and petechial purpura. The patient had fever and joint arthralgia. A skin biopsy showed leukocytoclastic vasculitis with IgA deposits on direct immunofluorescence. The diagnosis of immunoglobulin A vasculitis with annular leukocytoclastic vasculitis was made. The patient showed global improvement with topical steroids without relapse.
Conclusion: An annular variant of leukocytoclastic vasculitis is a rare manifestation of immunoglobulin A vasculitis.
|
Views: 1050
HTML: 24
PDF: 154
|
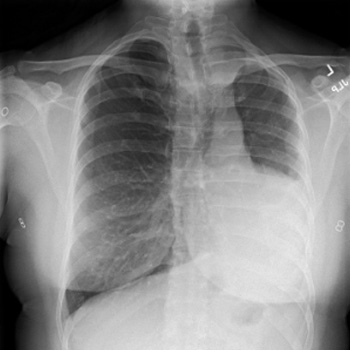
We report a rare yet successful utilisation of anti-CD20 therapy using rituximab for treatment of a case of IgG4-related mastitis proven by clinical, serological, and histopathological evidence. This was affecting a mid-aged female who was referred to the rheumatology clinic by the breast surgeons to help assessing for the possibility of an underlying inflammatory process involving the breast tissue unilaterally.
The clinical course was apparently complex with an onset of an induration in the right lateral superior quadrant of the breast with mild discomfort and heaviness sensation. This increased over a course of 2 weeks before presentation to the general surgery clinic.
Subsequent investigations confirmed that the case was IgG4-related mastitis and a trial of steroids and disease modifying anti-rheumatic drugs (DMARDs) was partially helpful, but not to a full degree, mandating the utilisation of a more advanced mode of therapy, so rituximab was selected.
|
Views: 131
HTML: 16
PDF: 144
|
Lung underdevelopment is a rare congenital anomaly with variable clinical significance and presenting symptoms. It usually manifests during childhood. We present two cases of developmental lung anomaly subtypes and discuss clinical presentation and outcomes in such patient populations.
| 2.1 = | 1.762 Cit. to date |
| 842 Docs. to date |




Publisher
Official Journal of the
European Federation of Internal Medicine
www.efim.org
Publisher: SMC media Srl
Via Giovenale, 7 - 20136 Milan - Italy
P.IVA 07626490960
info@ejcrim.com
www.ejcrim.com - ISSN: 2284-2594 - © EFIM 2014-2024, Published by SMC Media srl, Italy - Privacy policy

