EJCRIM 2023 CiteScore
| 2.1 = | 1.762 Cit. to date |
| 842 Docs. to date |
Last updated on 05 May, 2024
Updated monthly
Updated monthly
Powered by



|
Views: 0
HTML: 0
PDF: 0
|
Background: Arteriovenous malformations (AVMs) are abnormal direct connections between arterial and venous systems, without an interposed capillary bed. This permits high-flow arteriovenous shunting, which precipitates structural changes in the afferent and efferent vessels, namely arterial smooth muscle hyperplasia and thinning of venous walls. Patients with intracranial AVMs typically present with a haemorrhage, headache or seizure. Treatment is either via medical management aimed at control of seizures, headache and blood pressure, or interventional via surgical, radiation or radiologically guided embolisation.
Case description: We report the case of a woman in her early 40s presenting with a tonic-clonic seizure against a background of a 31-year history of migraine and an 18-month history of tremors in her right arm. The clinical examination was remarkable for an extremely loud cranial bruit and a right homonymous hemianopia. Imaging diagnosed an 8 cm Martin-Spetzler grade V intracranial arteriovenous malformation in her left parietal lobe, which was deemed unsuitable for operative or radiotherapy-based intervention.
Conclusion: The patient was managed through observation and relatively good control of her breakthrough seizures was achieved through the addition of brivaracetam to her lamotrigine and carbamazepine-based therapy, six years after her initial presentation.

|
Views: 21
HTML: 0
PDF: 3
|
Background: Hypertrophic pachymeningitis (HP) is a disease with diverse aetiologies, including the autoimmune one, either associated with antineutrophil cytoplasmic antibodies or immunoglobulin G4.
Case description: A 65-year-old woman with a history of systemic arterial hypertension, presented with intense progressive headaches. HP and hemispheric vasogenic oedema were observed by nuclear magnetic resonance (NMR) study. During the six months before the headache, she had developed progressive hearing loss which she attributed to age. A biopsy of dura mater showed necrotising vasculitis with peripheral inflammatory infiltrate, made up of accumulations of epithelioid cells and multinucleated giant cells, and abundant eosinophils. A final diagnosis of HP with eosinophilic granulomatosis with polyangiitis (EGPA) was made.
Discussion: The patient had eosinophilic granulomatosis with polyangiitis (EGPA) histology, ANCA-negative serology and HP. This case is important because it shows that EGPA seems to have a spectrum of clinical diseases, including HP with negative serology, and bilateral sensorineural hearing loss.
Conclusion: We are facing a wide spectrum of EGPA, breaking the paradigm of only systemic involvement.

|
Views: 25
PDF: 19
HTML: 6
|
Background: Anti-leucine-rich glioma inactivated 1 limbic encephalitis (anti-LGI1 LE) is one of the most frequent autoimmune encephalitis, commonly coexisting with other autoimmune diseases. Rheumatoid arthritis (RA) and monoclonal gammopathy of unknown significance (MGUS) are commonly associated with autoimmune phenomena. However, neither RA nor MGUS have been described in the literature to date as coexisting with anti-LGI1 LE.
Case description: We present the case of anti-LGI1 LE in a male patient with rheumatoid arthritis, who was also found to have an MGUS. The patient was initially treated with corticosteroids and IV immunoglobulin. After a mild relapse, his treatment was complemented with rituximab, resulting in complete regression of the disease symptoms.
Conclusions: Our report provides evidence for the coexistence of anti-LGI1 LE with RA and/or MGUS, thus extending the differential diagnosis of patients suffering with these disease entities that present with neuropsychiatric symptoms suggestive of encephalitis. Moreover, this case raises challenges on the management of the coexistence of these diseases, given the lack of therapeutic guidelines and their potential interaction on a pathophysiological and a clinical level.

|
Views: 42
HTML: 6
PDF: 24
|
Background: We describe a case of myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) in a 16-year-old patient who initially presented with clinical features of septic meningoencephalitis. This case outlines the importance of considering a diagnosis of MOGAD in patients who fail to improve with appropriate antimicrobial therapy or show a positive clinical response to glucocorticoids (often used in treatment of meningococcal meningitis). We emphasise the importance of recognising that an infectious prodrome can precede MOGAD.
Case description: A 16-year-old male was admitted with vomiting, fever, headache, photophobia and altered mental state. He was treated for meningoencephalitis with initial clinical improvement. Lumbar puncture findings were suggestive of viral meningoencephalitis. During admission the patient went through several periods of transient clinical and biochemical improvement, alternating with periods of symptomatic relapse. On day 17 of admission, he was transferred to a tertiary centre for suspected autoimmune disseminated meningoencephalitis (ADEM) and two days later, he suffered a catastrophic neurological decline with new dysarthria, dysphagia, aphasia, horizontal nystagmus and facial paralysis. He made a remarkable neurological recovery after commencing treatment with IV immunoglobulin, IV methylprednisolone and plasma exchange, with complete resolution of symptoms.
Conclusion: MOGAD can run a variable course and present soon after a central nervous system infection, making the diagnosis more challenging. Nonetheless, patients can achieve a full neurological recovery with early recognition, diagnosis and treatment of this rare entity.

|
Views: 56
HTML: 9
PDF: 42
|

|
Views: 214
HTML: 11
PDF: 110
|
Bronchial artery embolization (BAE) is a procedure that aims to control bleeding from bronchial arteries in massive and chronic haemoptysis. It is considered to be a life-saving measure in severe life-threating haemoptysis. Although it is minimally invasive and has a high success rate, it still carries a list of complications. These include post-embolisation syndrome, chest pain, back pain, dysphagia, vascular injury at the site of the embolisation leading to dissection, perforation, pseudoaneurysm and, very rarely, embolic infarction to non-target vessels.
Stroke is one of the rare complications post BAE, and it can be severe and fatal. Few cases of stroke post BAE have been reported in the literature, and they were mainly due to posterior cerebral circulation infarction. Here, we report a case of a stroke post BAE due to massive middle cerebral artery (MCA) infarction and to our knowledge it seems to be the first reported case of MCA infarction post BAE.
The discussion will cover the possible mechanisms of embolic passage, the outcome of the case including rehabilitation perspectives and the learning points. These will also highlight the importance of early recognition, which can save patients from a disabling stroke in the future.
|
Views: 185
HTML: 9
PDF: 153
|
Background: Hyperargininemia is a rare inherited metabolic disorder of the urea cycle with an autosomal recessive transmission. It occurs due to a deficiency of the enzyme arginase I and causes progressive neurological damage. Very few cases are diagnosed in adulthood, with the majority being diagnosed before the age of 4. Currently, this condition is diagnosed by a mass spectrometry technique in neonatal screening, which has been implemented in Portugal since 2007; births before that were not screened for this entity.
Case description: We present a case of a 23-year-old woman referred to the internal medicine and neurology departments with a history of two hospital admissions for rhabdomyolysis at the age of 18, consanguineous parents, learning difficulties and multiple falls since the age of 8. In addition, the patient also had behavioural changes so she had psychological counselling at school, but lacked family support. Neurological examination showed mild proximal paraparesis, and spastic and paraparetic gait. The aetiological study revealed a pathological variant in homozygosity ARG1 and increased blood levels of arginine. Therefore, the diagnosis of hyperargininemia was confirmed.
Conclusions: Compared to other urea cycle disorders, hyperargininemia is the rarest one. It is important to recognise the characteristic clinical features and diagnose it early because a favourable outcome can be achieved with appropriate treatment. This case shows a delayed diagnosis of hyperargininemia and highlights the importance of the internist’s role in diagnosing rare diseases.
|
Views: 212
HTML: 31
PDF: 164
|
We present a 30-year-old male who sustained a mild traumatic brain injury and then was intubated due to deterioration of consciousness. A head CT scan revealed mild brain oedema, a fractured nasal bone and mild left thoracic wall haematoma. Despite complete clinical and radiological normalisation within 36 hours, he failed to wean off the ventilator. The patient was found to have subtle bulbar manifestations including dysphonia, dysarthria, and dysphagia, with recurrent left lung collapse. He responded to an empirical pyridostigmine trial despite negative biochemical tests for myasthenia gravis (MG). The patient was weaned successfully from the ventilator, transferred to a long-term care facility, and then discharged home. Classic symptoms and signs of a disease may be absent, but the presence of dysarthria, dysphagia, transient vocal cord palsy, nasal speech, absent gag reflex and respiratory failure in difficult-to-wean patients, with no definitive diagnosis, may warrant an empirical trial of therapy for suspected MG and for the benefit of any doubt.
|
Views: 276
HTML: 35
PDF: 314
|
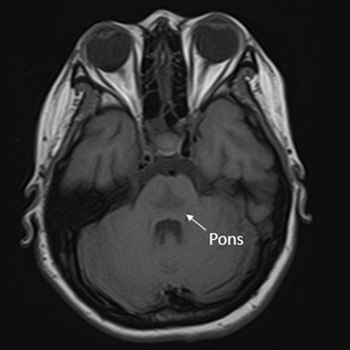
Osmotic demyelination syndrome (ODS) is a disorder characterised by the widespread development of demyelination in both pontine and extrapontine regions. It has been recognised as a complication arising from the rapid correction of hyponatraemia. This study presents the case of a 20-year-old Thai female patient at 10 weeks gestation, exhibiting an initial presentation of catatonia – an uncommon manifestation of ODS. The patient developed symptoms following the rapid correction of hyponatraemia in the context of hyperemesis gravidarum. Magnetic resonance imaging (MRI) of the brain revealed a trident or bat-wing-shaped pattern in T2-weighted and fluid-attenuated inversion recovery (FLAIR) sequences at the central pons. The patient underwent five cycles of plasmapheresis and received rehabilitation, leading to clinical improvement.

|
Views: 179
HTML: 20
PDF: 174
|
| 2.1 = | 1.762 Cit. to date |
| 842 Docs. to date |




Publisher
Official Journal of the
European Federation of Internal Medicine
www.efim.org
Publisher: SMC media Srl
Via Giovenale, 7 - 20136 Milan - Italy
P.IVA 07626490960
info@ejcrim.com
www.ejcrim.com - ISSN: 2284-2594 - © EFIM 2014-2024, Published by SMC Media srl, Italy - Privacy policy

