EJCRIM 2023 CiteScore
| 2.1 = | 1.762 Cit. to date |
| 842 Docs. to date |
Last updated on 05 May, 2024
Updated monthly
Updated monthly
Powered by



|
Views: 0
HTML: 0
PDF: 0
|
Takayasu arteritis (TA) primarily causes ischaemic nephrosclerosis but can occasionally be associated with glomerulopathy. We report a case of a female in her twenties with PLA2-negative, THSD7A-positive membranous nephropathy (MN) refractory to rituximab, who presented with neck pain and new-onset hypertension. Blood work showed elevated inflammatory markers. Imaging of the head and neck revealed focal dilation and irregularity of the vertebral arteries, consistent with TA. The patient was started on treatment with steroids, followed by mycophenolate mofetil, which led to the resolution of symptoms and nephrotic syndrome. This case highlights an uncommon sequence of events, with MN presenting before TA, underscoring the need to consider TA in differentials for patients with MN. Notably, this is the first reported case in a young female, emphasising the need for further understanding of TA-associated glomerular diseases. Additionally, the presence of THSD7A in MN, despite negative malignancy workup, is also noteworthy.

|
Views: 4
HTML: 0
PDF: 1
|
A 50-year-old patient with a history of limited cutaneous scleroderma began with polyarthralgia (left shoulder, elbows and hips) without stiffness or associated inflammatory syndrome. Treatment with oral anti-inflammatory drugs was started on suspicion of peripheral spondyloarthritis with partial response. This progressed with the appearance of stiffness and functional limitation of the hips as well as an increase in the inflammatory syndrome two weeks after onset. It was decided to perform an 18F-FDG-PET scan compatible with polymyalgia rheumatica. The patient was treated with oral corticosteroids with an excellent response after one week of treatment.

|
Views: 39
HTML: 6
PDF: 18
|
Introduction: Polymyalgia rheumatica (PMR) is a chronic inflammatory disorder that causes stiffness and pain in the proximal joints, including the shoulders, hips and neck. The exact cause of polymyalgia rheumatica is yet to be fully understood, but research suggests that both genetic and environmental factors may contribute to it. Studies have previously linked the onset and relapse of polymyalgia rheumatica symptoms to the influenza and COVID-19 vaccines. The Food and Drug Administration approved the respiratory syncytial virus (RSV) vaccine, which is a recombinant protein vaccine for adults over 60, in May 2023. No previous reports of polymyalgia rheumatica onset or relapse have been linked to the RSV vaccine. The human proteome shares some peptides with the RSV F antigen, suggesting a high risk of cross-reactivity when using that antigen in vaccination formulations.
Case description: A 72-year-old man experienced a new onset of bilateral shoulder pain and stiffness three days after receiving the Abrysvo® RSV vaccine. The symptoms lasted more than an hour (up until noon) and interfered with his activities of daily living. Inflammatory markers such as C-reactive protein were elevated. The patient’s symptoms and inflammatory marker levels significantly improved with prednisone therapy.
Conclusion: In patients with typical PMR symptoms, it is important for clinicians to carefully review immunisation history to rule out any potentially related adverse effects.

|
Views: 25
PDF: 19
HTML: 6
|
Background: Anti-leucine-rich glioma inactivated 1 limbic encephalitis (anti-LGI1 LE) is one of the most frequent autoimmune encephalitis, commonly coexisting with other autoimmune diseases. Rheumatoid arthritis (RA) and monoclonal gammopathy of unknown significance (MGUS) are commonly associated with autoimmune phenomena. However, neither RA nor MGUS have been described in the literature to date as coexisting with anti-LGI1 LE.
Case description: We present the case of anti-LGI1 LE in a male patient with rheumatoid arthritis, who was also found to have an MGUS. The patient was initially treated with corticosteroids and IV immunoglobulin. After a mild relapse, his treatment was complemented with rituximab, resulting in complete regression of the disease symptoms.
Conclusions: Our report provides evidence for the coexistence of anti-LGI1 LE with RA and/or MGUS, thus extending the differential diagnosis of patients suffering with these disease entities that present with neuropsychiatric symptoms suggestive of encephalitis. Moreover, this case raises challenges on the management of the coexistence of these diseases, given the lack of therapeutic guidelines and their potential interaction on a pathophysiological and a clinical level.

|
Views: 53
HTML: 3
PDF: 44
|
Background: The psychiatric manifestations of Sjögren’s syndrome are often overlooked despite their prevalence. They can be revelatory of the disease and include anxiety, depression, dementia and, rarely, psychosis.
Case description: We report a case of 18-year-old female in whom a major depressive syndrome revealed primary Sjögren’s disease, with a favourable outcome after treatment with rituximab.
Conclusion: The diagnostic of Sjögren’s syndrome should be considered in patients who present with unexplained and refractory neuropsychiatric symptoms, even in the absence of sicca symptoms.

|
Views: 56
HTML: 9
PDF: 42
|

|
Views: 312
HTML: 64
PDF: 167
|
The incidence of post-infectious autoimmune diseases has been on the rise following the COVID-19 pandemic. Recently, an autistic patient was admitted to the hospital presenting with a mild upper respiratory system COVID-19 infection. Months after recovery and polymerase chain reaction negativity, the patient developed HEp-2 cell positivity and presented with relapsing polychondritis (RP), a rare autoimmune disease. The mechanism of this autoimmune invasion is ultimately caused by activating a myriad of immune reactions. Lymphocytopenia almost always accompanies various clinical forms of COVID-19; however, it may drive the lymphocytopenia-induced proliferation of autoreactive T cells via the activation of interleukin-6 (IL-6). Moreover, high levels of neutrophils during infection promote autoimmune disease by releasing cytokine and chemokine cascades that accompany inflammation, and neutrophil extracellular traps regulating immune responses through cell–cell interactions. Furthermore, autism spectrum disorder patients display an altered immune system that includes an augmented inflammatory cytokine milieu leading to an increased pro-inflammatory Th1/Th2 ratio. In addition, the pathophysiology of RP is majorly associated with a cell-mediated immune reaction; thus, the predisposing exaggerated immune system of such patients must also be considered as a predisposing factor to the development of post-infectious autoimmune diseases.

|
Views: 288
HTML: 14
PDF: 140
|
Background: Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease, characterised by multi-organ affections. Haematological involvement is a common manifestation of SLE, consisting of autoimmune peripheral cytopenia. Autoimmune myelofibrosis (AIMF) is a rare cause of cytopenia in SLE; it could precede or be concurrent with the diagnosis of SLE. There are few studies that describe this association.
Case description: We report a case of AIMF revealing the diagnosis of SLE in a 34-year-old female, presented with episodes of gingival bleeding associated with peripheral inflammatory polyarthralgia, photosensitivity and deterioration of general condition. Clinical examination revealed a soft pitting oedema in the lower limbs. Laboratory investigations showed a pancytopenia, inflammatory biological syndrome, with positive 24-hour proteinuria and anti-native DNA antibodies. A bone marrow biopsy showed diffuse myelofibrosis associated with maturation disorders and no tumour infiltrate. Renal biopsy revealed proliferative glomerulonephritis class III with immune deposits.
Conclusion: The association of AIMF with SLE has been rarely reported, and it could be another cause for cytopenia in SLE.
|
Views: 137
PDF: 121
HTML: 14
|
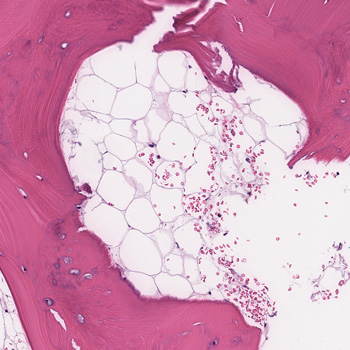
Intracoronary in-stent restenosis (ISR) is a phenomenon that generally occurs between 3 and 6 months after stent placement. With the introduction of drug-eluting stents (DES), the incidence of ISR has decreased but not disappeared. We report a case of reiterant in-stent restenosis of an 81-year-old female patient who underwent multiple percutaneous coronary intervention and two coronary artery bypass surgeries. ISR is possibly associated with extra-stent, stent-related and intra-stent factors. Here, we excluded the first two and focused on the intra-stent factors that seem more likely in our case. A challenging diagnostic workup led us to the hypothesis of a coronary vasculitis potentially triggered by some component of the stent in a predisposed patient carrier of non-disease-specific ANA, with an exaggerated immune response. No recurrence of ISR occurred after the introduction of steroids. Biological and intra-stent causes of ISR should be taken into careful consideration to aim for the early detection of the underlying mechanism of restenosis and to embrace the best therapeutic strategy.
|
Views: 718
HTML: 45
PDF: 431
|
Background: Autoimmune diseases are not contraindications for immune checkpoint inhibitors (ICI) therapy in patients with cancer. However, immune-related adverse events (irAEs) are frequently observed in patients receiving ICIs including dermatitis, thyroiditis, colitis, and pneumonitis. Thrombocytopenic purpura, aplasia, and haemophagocytic lymphohistiocytosis (HLH) are rarely observed during ICIs.
Case description: We report the case of a male patient with pre-existing untreated HLA B27 and ankylosing spondylitis with gastric cancer and liver metastases. The 79-year-old man was treated with anti-HER2 trastuzumab and anti-PD-1 nivolumab. Seventeen days after the seventh cycle of treatment, he presented at the emergency department with acute fever, confusion, and hypotension. Laboratory results showed pancytopenia, and elevation of ferritin and triglyceride. No infections were detected. Although not seen in a bone marrow biopsy, clinical presentation, and absence of infection, together with an H-score of 263, indicated HLH. The patient was treated with dexamethasone for four days and discharged on a tapering dose of steroids. At the two-month follow-up, clinical presentation was normal and blood test almost normalised. At 8 months, no liver metastases were observed.
Conclusions: In a patient with a pre-existing autoimmune condition, immunotherapy led to the development of HLH, which was controlled by glucocorticoid. Absence of the feature of haemophagocytosis in the bone marrow biopsy did not exclude the diagnosis, as HLH can occur in the spleen or in the liver. Glucocorticoid therapy did not prevent the anti-cancer effect of ICIs, and liver metastases disappeared 8 months post-HLH. This case warrants further research on the interplay between autoimmunity and ICI response, as well as ICI-induced irAEs.
| 2.1 = | 1.762 Cit. to date |
| 842 Docs. to date |




Publisher
Official Journal of the
European Federation of Internal Medicine
www.efim.org
Publisher: SMC media Srl
Via Giovenale, 7 - 20136 Milan - Italy
P.IVA 07626490960
info@ejcrim.com
www.ejcrim.com - ISSN: 2284-2594 - © EFIM 2014-2024, Published by SMC Media srl, Italy - Privacy policy

