Keywords
Caroli disease, polycystic kidney disease, hepatomegaly, intrahepatic bile ducts
Abstract
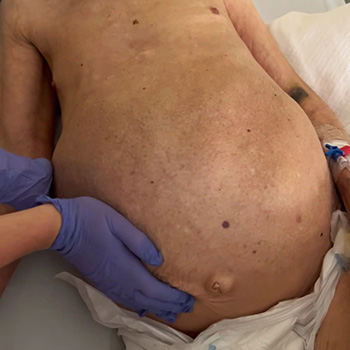
Caroli disease is a rare congenital pathology caused by mutation of the PKHD1 gene (polycystic kidney and hepatic disease 1), also responsible for autosomal recessive polycystic kidney disease. Characterized by segmental and multifocal dilatation of the large intrahepatic bile ducts, classic disease involves only malformation of the biliary tract. The association with congenital hepatic fibrosis is called Caroli syndrome. We describe the case of an 84-year-old man with Caroli syndrome diagnosed in 1997 by liver biopsy. The CT scan revealed massive hepatomegaly, extending to the pelvic region, and almost total replacement of the parenchyma by numerous cystic formations, no evidence of bile duct dilatation, and no ascites or splenomegaly suggestive of portal hypertension. The atypical clinical presentation, with no reported complications, resembles that of a space-occupying lesion with an indolent course, previously misdiagnosed as metastatic neoplasm.
References
Views: 419
HTML downloads: 98
PDF downloads: 340
Published:
2023-03-18
Issue:
2023: Vol 10 No 3
(view)






