
Keywords
Neurofibromatosis type 1, epithelioid sarcoma, soft-tissue tumour
Abstract
Background: Patients with neurofibromatosis type I (NF1) have an increased risk of developing soft-tissue sarcomas, particularly those related to the nervous system. Epithelioid sarcoma (ES) is an exceptionally rare subtype of soft-tissue sarcoma, with limited knowledge about its clinical presentation and optimal management in NF1. This report aims to provide insights into the characteristics and outcomes of ES in NF1 patients.
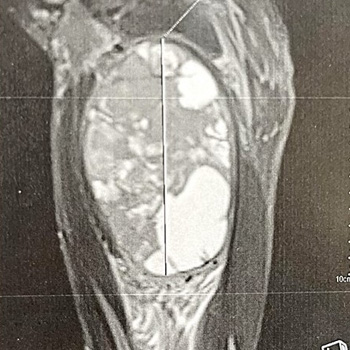
Case description: A 37-year-old man with a history of NF1 presented with a progressively worsening mass on his right inner thigh. An MRI scan revealed a well-defined tissue mass originating from the adductor magnus muscle, later confirmed as ES through histopathology and immunohistochemistry. Considering poor local and general prognosis, the multidisciplinary team recommended salvage hip disarticulation, however the patient refused and opted for palliative marginal resection to reduce the tumour size. The patient’s condition declined rapidly, and he succumbed six days after the surgery.
Conclusion: This case highlights the rarity of ES in NF1 patients and underscores the potential for malignant tumour development in this population. Further research is needed to improve our understanding and management of sarcomas in the context of NF1.
References
Views: 103
HTML downloads: 11
PDF downloads: 58
Published:
2024-03-28
Issue:
2024: Vol 11 No 4
(view)






