EJCRIM 2023 CiteScore
| 2.1 = | 1.730 Cit. to date |
| 842 Docs. to date |
Last updated on 05 March, 2024
Updated monthly
Updated monthly
Powered by




|
Views: 1828
HTML: 3848
PDF: 422
|
Modern medicine began in the last half of the nineteenth century when doctors started practising the scientific method at the bedside. However, in his presidential address to the Association of American Physicians in 1979 James Wyngaarden postulated that the clinical scientist was an endangered species. Several reasons for this have been suggested, including “the seductive incomes that now derive from procedure-based specialty medicine”. Others have suggested that it is simply because the things left to be discovered at bedside have become exhausted, and that all the big medical advances will now be made by high-powered institutions.
|
Views: 1367
HTML: 653
PDF: 748
Authors Information: 0
Figure and Table: 0
References: 0
Copyright authorization form: 0
Letter to the editors: 0
Conflicts of interest disclosure: 0
|
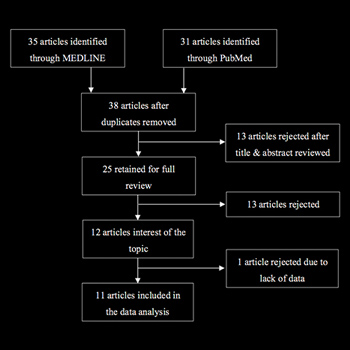
Objectives: To perform a literature review and estimate MG incidence in an SLE cohort.
Materials and methods: We searched MEDLINE and PubMed for case reports of SLE and MG. We also calculated MG incidence within our clinical SLE cohort (females only).
Results: Eleven articles met our criteria, providing 13 SLE patients who developed MG. The majority were female (84.6%), with the average ages of 25.6 and 33.5 years at diagnoses of SLE and MG, respectively. In 380 SLE female patients followed for 2,850 person-years, one MG case occurred
Conclusion: MG in SLE is a rare event.

|
Views: 1297
HTML: 1962
PDF: 745
Cover Letter: 0
Tables: 0
Copyright: 0
Consent 1: 0
Consent 2: 0
Consent 3: 0
consent 4: 0
Consent 5: 0
Tables after correction: 0
LEARNING POINTS: 0
|
A 41-year-old man was admitted to an intensive care unit following respiratory arrest. One day prior to admission, he had complaints of nausea and pain involving lower limbs. On the night of admission he developed diplopia, dysphagia, and rapidly progressive quadriparesis. He developed respiratory failure requiring mechanical lung ventilation 24 hours later. On the fifth day of hospital stay the patient became comatose with absent brainstem reflexes and appeared to be brain dead. The cerebrospinal fluid showed albuminocytological dissociation. The electroencephalogram revealed an alpha rhythmical activity. The electrophysiological evaluation revealed an inexcitability of all nerves. Guillain-Barré syndrome was suspected. With supportive treatment the patient had a remarkable recovery and now is able to independently conduct his daily activities.
|
Views: 830
HTML: 451
PDF: 396
Untitled: 0
figure: 0
figure legend: 0
conflicts of interest: 0
copyright form: 0
Breast carcinoma with choriocarcinomatous differentiation; a rare case (Choriocarcinomatous breast carcinoma): 0
|
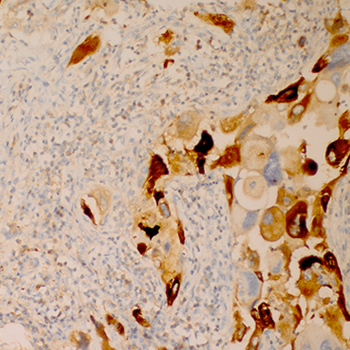
Objectives: Breast carcinoma with choriocarcinomatous differentiation is a rare entity, generally presenting with high-grade disease and an aggressive clinicalcourse with overall survival of less than a year
Case: A 69-year-old woman with a diagnosis of pT1N0M0 invasive ductal carcinoma with choriocarcinomatous differentiation received six cycles ofadjuvant chemotherapy and is still disease free on the 23rd month after diagnosis, showing a better prognosis than most other cases reported in theliterature.
Conclusion: The reason for the poor prognosis for this type of breast carcinoma remains unclear. Standard chemotherapeutic agents administered for breastcarcinoma may be used for choriocarcinomatous differentiation.
|
Views: 789
HTML: 444
PDF: 448
|
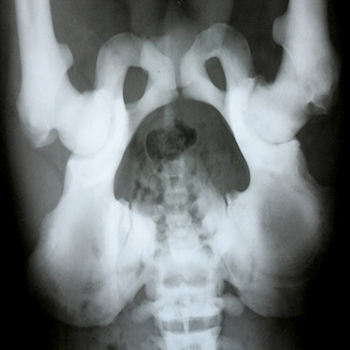
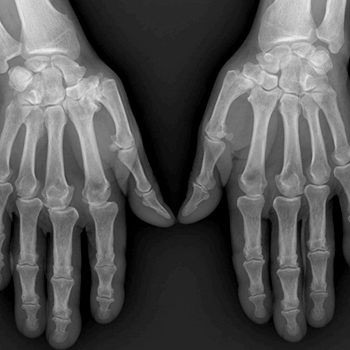
Objectives: Autosomal dominant osteopetrosis (ADO) is a rare genetic disease characterized by increased bone mass and density due todefective bone resorption. The aim of this case study is to present the clinical and radiographic features of a 22-year-old male patient with ADO andto serve as a reminder that this rare disease should be considered in the differential diagnosis of chronic low back pain.
Materials and methods: A 22-year-old patient with ADO is presented in this case report.
Results: Clinical and radiographic features of the patient were consistent with ADO.
Conclusions: ADO should be taken into consideration in differential diagnosis of low back pain.
|
Views: 1474
HTML: 1752
PDF: 366
Fig. 1: 0
Fig. 2: 0
Copyright Authorization: 0
Conflict of Interests: 0
|
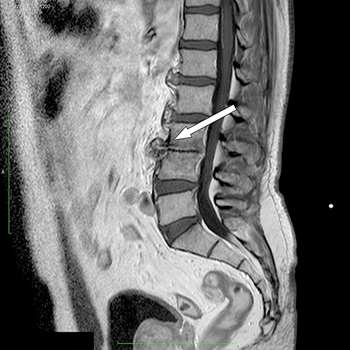
A 61-year-old man presented with high fever, and severe back and abdominal pain following transrectal ultrasonography (TRUS)-guided prostate biopsy. Diagnosis of spondylodiscitis and psoas abscesses was made based on MRI images of the lumbar tract of the spine. Six-monthbroad-spectrum antibiotic treatment and immobilization with a girdle overcame the disease without any relapse at the 1-year follow-up.
Spondylodiscitis after TRUS-guided prostate biopsy is a rare event, which is not yet included as a major complication of the procedure. It is probably due to thepresence of fluoroquinolone-resistant bacteria in faeces. It is, therefore, important to highlight this possibility and to stressthe use of targeted antibiotic prophylaxis after rectal flora swabbing with selected antibiotics at sufficient concentrations to be effective.

|
Views: 992
HTML: 345
PDF: 361
Table 1: Cases of onset uveitis under TNF blockers: 0
Bilateral panuveitis at etanercept initiation for Juvenile idiopathic arthritis: 0
Bilateral panuveitis at etanercept initiation for Juvenile idiopathic arthritis: 0
Bilateral panuveitis at etanercept initiation for Juvenile idiopathic arthritis: 0
Conflicts of interest: 0
copyright authorizaion form: 0
|
Introduction: Uveitis is a well-known extra-rheumatological manifestation of juvenile idiopathic arthritis (JIA). Tumour necrosis factor(TNF) has been used to treat uveitis associated with inflammatory diseases. A new-onset uveitis under anti-TNF therapy is uncommon.
Case presentation: A 12-year-old male, affected since the age of 6 years, by a severe form of polyarticular JIA. When etanercept was started, hepresented panuveitis bilaterally, so we switched to infliximab with good response.
Conclusions: The TNF-soluble receptor could be considered as a possible promoter in inducing endogenous new-onset uveitis in JIA.
|
Views: 921
HTML: 747
PDF: 1959
cover letter: 0
cover letter: 0
Figure 1: 0
Figure 2: 0
Conflicts of Interests Disclosure 1: 0
Conflicts of Interest Disclosure 2: 0
Conflicts of Interest Disclosure 3: 0
Copyright: 0
|
Arnold–Chiari malformation is defined as downward displacement of the brainstem and cerebellum through the foramen magnum. It hasdifferent clinical presentations and four subtypes. It is known that downward migration of posterior fossa components through the foramen magnum and associated lower cranial nerve palsy and brainstem compression can cause respiratory failure. Acute respiratory failure could mark the onset of thedisease. Posterior fossa decompression performed to treat primary disease can improve the central sleep abnormalities. As respiratory failure is rarely seen, this paper presents two cases of Arnold–Chiari malformation with respiratory failure.
|
Views: 1121
HTML: 626
PDF: 456
Figure 1: 0
Table 1: 0
Author Information Page: 0
Conflict of Interests Disclosure: 0
Copyright Transfer Declaration: 0
Ethical Approval: 0
Original File: 0
|
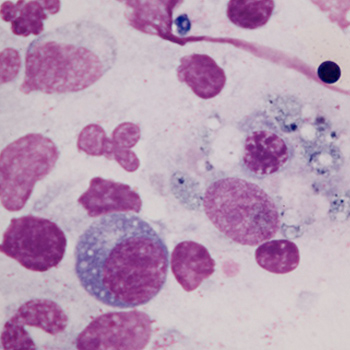
We describe a 62-year-old patient with a 4-year history of myelodysplasia who later developedstriking features that included massive splenomegaly, rapidly evolving visual loss and a sensorimotor polyneuropathy. This led us to consider the diagnosisof haemophagocytic lymphohistiocytosis (HLH). Upon further investigation, we found that he fulfilled the necessarydiagnostic criteria for HLH, including the presence of haemophagocytosis of erythroid precursors on bone marrow smear.

|
Views: 911
HTML: 713
PDF: 896
Abstract: 0
Authorsinformation: 0
Conflicts of Interest Dr van Aken: 0
Conflicts of interest Dr van Nieuwkoop: 0
Conflicts of Interest Drs Vlasveld: 0
Copyright of Authorization form: 0
Figure1: 0
Informed consent patient: 0
List references: 0
Table1: 0
|
Objectives: To illustrate that the protease inhibitor (PI) ritonavir, widely used as part of the treatment for HIV, might cause drug–drug interactions with inhaled corticosteroids.
Material and methods: A case report is presented.
Results: An HIV-positive patient presented with gradually changing body composition that was ascribed to lipodystrophy. Finally, iatrogenic Cushing's syndrome with secondary adrenal insufficiency was diagnosed due to a drug–drug interaction of ritonavir and fluticasone.
Conclusion: Lipodystrophy might mimic Cushing's syndrome. The combination of ritonavir and inhaled fluticasone may lead to systemic steroid excess causing Cushing's syndrome and secondary adrenal insufficiency.
|
Views: 1189
HTML: 461
PDF: 385
Fig 1: 0
Fig 2: 0
|
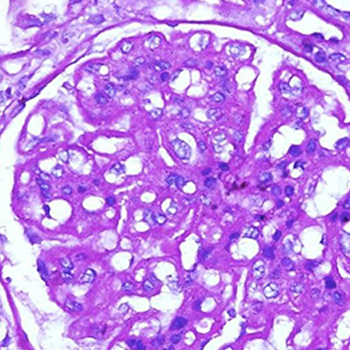
We present a rare case of a 23-year-old male incidentally detected with hepatitis B virus (HBV) infectionpresenting with features suggestive of HBV-associated nephropathy. A renal biopsy specimen suggested a mesangioproliferative glomerulonephritis with a full-house pattern on immunoflourescence consistent with a diagnosis of diffuse lupus nephritis. Glomerular HbeAg and HbsAg antigens were not detectable by immunofluorescence. Antiviral therapy was instituted to suppressviral replication, thereby leading to clinical and virological remission, including that of the glomerulonephritis, without the need for additional immunosuppressant therapy. This case depicts the uniqueness of the presentation of the two conditionsmimicking each other, the strategy adopted to prevent the activation of viral replication and the achievement of clinical remission.

|
Views: 727
HTML: 188
PDF: 353
Figure 1: 0
|
We present a case of an old woman with previously documented heparin-induced thrombocytopenia (HIT),treated with fondaparinux, who presented with thrombocytopenia and venous thrombosis after exposure to a preventive dose of fondaparinux duringorthopaedic surgery.
Any accidental exposure to heparin was avoided. Other causes of thrombocytopenia were excluded andantigenic tests combined with clinical probability made a diagnosis of HIT likely.
Can this be considered a possible case of fondaparinux-related HIT, despite the intense and earlydecrease in platelets, as usually happens in rapid-onset HIT, and the fact that previous exposure to fondaparinux had occurred 5 months previously?
|
Views: 1491
HTML: 1202
PDF: 590
Figure 1 showing purpura fulminans leading to peripheral gangrene in the feet.docx: 0
Figure 2 showing purpura fulminans leading to peripheral gangrene in the hands.docx: 0
Figure 3 A peripheral blood smear showing intracellular bacilli in neutrophils.docx: 0
Table 1 sepsis: 0
Copyright form: 0
|
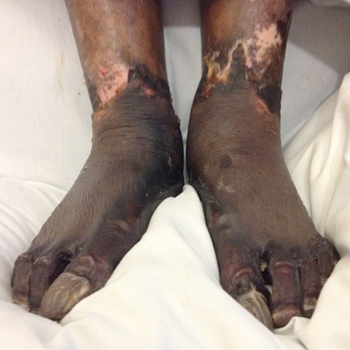
Infectious purpura fulminans is a rapidly progressive skin necrosis that has a mortality rate of 30%1,2. Here, we describe a case of infectious purpura fulminans caused by Capnocytophaga, diagnosed by a blood film.
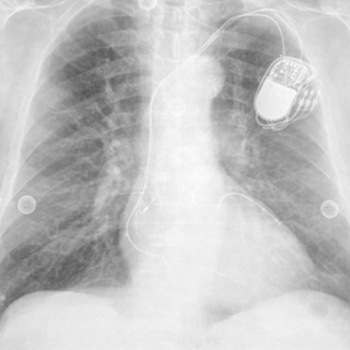
|
Views: 960
HTML: 975
PDF: 354
Author infomation page: 0
Figures: 0
Table: 0
Cover letter: 0
Copyright: 0
Disclosure: 0
|
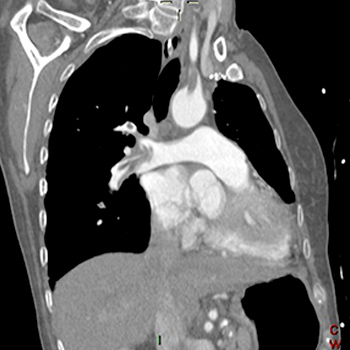
Objectives: We report a case of a frail older adult affected by Chilaiditi syndrome complicating transthoracic lung biopsy, which has never been presented before in the literature.
Materials and methods: After illustration of our case, we review the available literature about Chilaiditi syndrome.
Results: Chilaiditi syndrome is a rare disease characterised by the interposition of colonic segments between the liver and diaphragm associated with mild to severe clinical symptoms.
Conclusion: Although it is uncommon, Chilaiditi syndrome is a clinical condition that should be recognized early and differentiated from other diseases since it may significantly impair a patient's prognosis.
|
Views: 3207
HTML: 28538
PDF: 1087
Figure 1. Widened atrophic scarring and smooth and velvety skin, characterizing classical form EDS.: 0
|
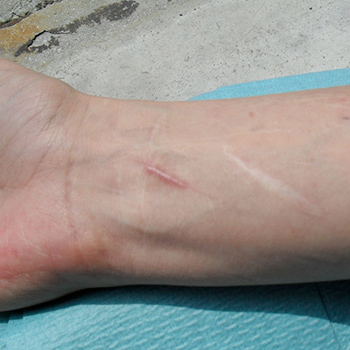
Ehlers–Danlos syndrome is a rare disease and a diagnostic challenge. This case report serves to remind the clinician that it is important to identify all affected patients in order to prevent complications.
|
Views: 749
HTML: 604
PDF: 423
Figure 1: 0
Figure 2: 0
Figure 3: 0
Figure 4: 0
Table 1: 0
|
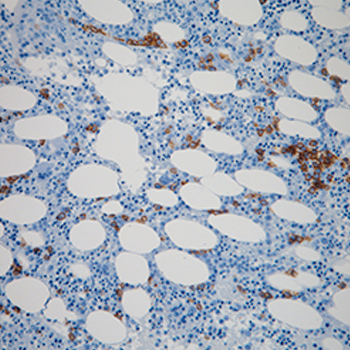
A 76-year-old man was admitted to hospital with fever, weight loss, pancytopenia, hepatosplenomegaly and a double monoclonal component IgM-IgG-k, suggesting a diagnosis of myeloma. Bone marrow and liver biopsies disclosed the presence of Donovan bodies, and the titre of anti-Leishmania antibodies was extremely high. After treatment with liposomal amphotericin B, the titre of antibodies fell considerably, while monoclonal components, pancytopenia and clinical symptoms slowly disappeared. Polyclonal ?-globulins are made of innumerable monoclonal components, one of which can appear as a recognizable band and be misdiagnosed as myeloma when representing the high titre of an antibody directed towards a specific antigen.
|
Views: 890
HTML: 282
PDF: 307
Conflicts of Interests + Copyright Authorization: 0
Figures: 0
|
Objectives: To present the possibility of acute arterial and venous thrombosis.
Materials and methods: Report of a patient presenting with acute dyspnoea and chest pain.
Results: Using a combined medical team and imaging studies, pulmonary embolism and acutearterial thrombosis were diagnosed. The patient was treated medically and surgically.
Conclusion: Physicians should be aware of the possibility of combined thrombosis and the diagnosis and management of the condition.

|
Views: 1177
HTML: 526
PDF: 379
Table 1: 0
AUTHOR INFORMATION PAGE: 0
Author's disclosure potential conflict of interests_Merlotti: 0
Author's disclosure potential conflict of interests_Carnevale: 0
Author's disclosure potential conflict of interests_Bellan: 0
Author's disclosure potential conflict of interests_Pirisi: 0
Author's disclosure potential conflict of interests_Rapetti: 0
EJCRIM_COPYRIGHT: 0
|
We report the case of an 86-year-old man with a past history of coronary disease admitted to ourinternal medicine department for severe asthenia and weakness due to rhabdomyolysis. Three days earlier, he had been discharged from a gastroenterology unit with adiagnosis of amoxicillin–clavulanate-induced acute cholestatic hepatitis. A review of his drugs revealed that he had taken atorvastatin 10 mg daily in theprevious six years, without clinical or laboratory signs of myopathy. Atorvastatin was therefore stopped, with gradual improvement of therhabdomyolysis. All concomitant drug therapy needs to be reassessed in elderly patients, especially when they become acutely ill.
|
Views: 1143
HTML: 276
PDF: 1251
diapositive1.tif: 0
CCF05052014.pdf: 0
reims: 0
|
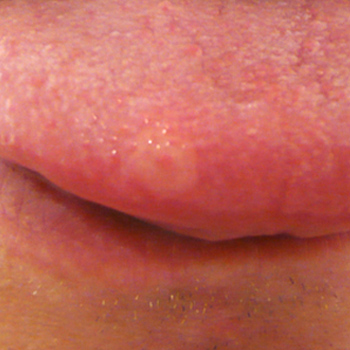
Objective: To report a case of Behc?et’s disease whose diagnosis was only confirmed thanks to an oral aphthous lesion biopsy.
Materials and methods: Conventional histopathological analysis of a biopsy of an aphthous oral lesion that had appeared two days previously.
Results: A small vein vasculitis with eosinophil and neutrophil granulocytes was evidenced.
Conclusion: The presence of a small vein vasculitis was here strongly in favour of Behc?et's disease, whereas such a diagnosis was not confirmed according to the International Study Group’s criteria.
|
Views: 1055
HTML: 297
PDF: 376
|
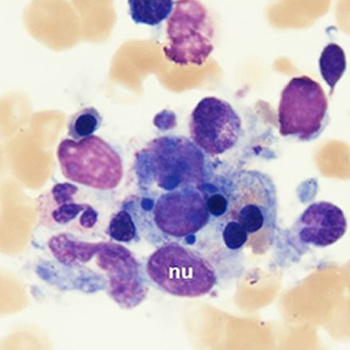
Objectives: Infectious agents triggering haemophagocyticlymphohistiocytosis (HLH) primarily involve the herpes virus group. We report a case of HLH precipitated by Plasmodium falciparum.
Materials and methods: Clinical and laboratory findings in a patient presenting with fever were collected. After confirmation of acute malaria, anti-malarial treatment was administered.
Results: Despite initial favourable evolution, the patient developed fever again together with a worsening of the haematological parameters and increased ferritin levels. A bone marrow biopsy confirmed the diagnosis of HLH.
Conclusion: This case illustrates that HLH should be considered in the differential diagnosis of acute malaria in patients with persisting fever and pancytopenia.
|
Views: 966
HTML: 414
PDF: 379
Figure 1: 0
Table 1: 0
Figure 2: 0
|
We report a case of a 55-year-old woman who was evaluated for multiple episodes of late postprandial hypoglycaemia. We diagnosed her condition as insulin autoimmune syndrome (Hirata disease) because of a high insulin autoantibody (IAA) titre in association with high levels of plasmatic insulin and hypoglycaemia in a patient with no history of exogenous insulin administration and the exclusion of other causes of late postprandial hypoglycaemia.

|
Views: 1781
HTML: 9715
PDF: 566
copyright burggraaff: 0
copyright linthorst: 0
author information: 0
copyright hoogerwerf: 0
Figure A: 0
Figure B: 0
|
Objectives: Pseudochromhidrosis is a rare condition where colours due to chromogenic microbial products or extrinsic chemicals are excreted with sweat. Chromhidrosis is the production of coloured sweat from apocrine or eccrine sweat glands. The aim of this case report is to illustrate all the steps involved in the diagnosis of pseudochromhidrosis.
Materials and methods: A 17-year-old patient with pseudochromhidrosis is presented.
Results: Clinical features of the patient were consistent with pseudochromhidrosis.
Conclusions: The distinction between chromhidrosis and pseudochromhidrosis can be made based on a detailed history, skin biopsy and empiric treatment.
|
Views: 690
HTML: 208
PDF: 295
Figure 1: 0
Figure 2: 0
Figure 3: 0
Author information page: 0
|
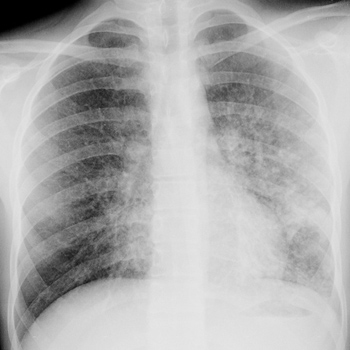
Objective: Interstitial lung diseases (ILD) are a group of pathologies of undetermined frequency that require a broad differential diagnosis and continue to pose a challenge for clinicians.
Observations: We present a clinical case of a 17-year-old male with acute interstitial pneumonitis, lung aspergillosis and foreign body lung granulomatosis after carbon monoxide (CO) intoxication. As far as we know, no similar cases have been reported in the literature.
Conclusions: ILD require a broad differential diagnosis, which is of great importance to prognosis.

|
Views: 838
HTML: 607
PDF: 288
Untitled: 0
Learning points+references: 0
|
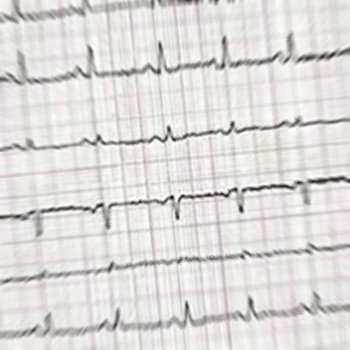
Objectives: Primary adrenal insufficiency AI is regarded as a progressive disease needing lifelong replacement therapy, but this may not always be the case.
Material and methods: A non-acute presentation of AI following a hypotensive episode caused by blood loss was investigated.
Results: Adrenal function fully recovered without treatment.
Conclusions: There should be a high index of suspicion and a low threshold for performing tests of adrenal function in survivors of critical illness and severe hypotensive episodes.
|
Views: 791
HTML: 432
PDF: 775
Figure 2: 0
Figure 1: 0
Cover Letter: 0
|
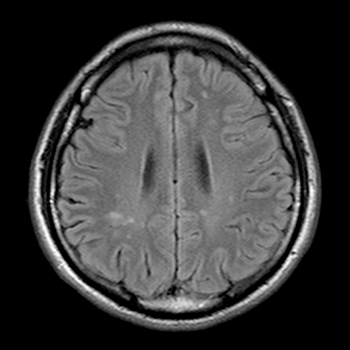
A 31-year-old man with pontine infarction was referred to our hospital for further evaluation and treatment. At admission, his neurological examination was unremarkable. No lymphadenopathy or skin lesions were found. The Treponema pallidum haemagglutination test, rapid plasma regain test and fluorescent treponemal antibody absorption test of immunoglobulin G were positive in both serum and cerebrospinal fluid (CSF). CSF analysis showed lymphocytic pleocytosis. The patient had male-to-male sexual contact and was found to be HIV positive. Physicians should be aware that acute ischaemic stroke may be the first manifestation of neurosyphilis in a young adult, especially with HIV infection.
|
Views: 1249
HTML: 382
PDF: 339
Figure 1a: 0
Figure 1b: 0
Figure 1c: 0
Figure 2a: 0
Figure 2b: 0
|
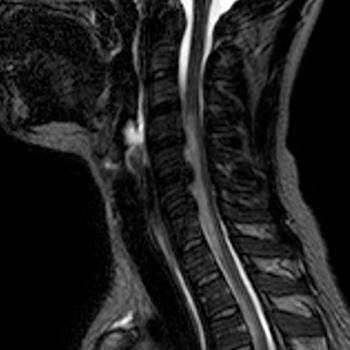
Several synkinesis syndromes have been reported in the literature. Synkinesis syndromes are rare and are most commonly congenital or follow post-traumatic reinnervation. We describe a novel synkinesis syndrome that developed several months after cervical spinal cord infarction due to a herniated disc in a 29-year-old woman. When the patient overstretched the extensor muscles of the right hand, the right upper eyelid raised automatically and nasal congestion developed. We hypothesize that aberrant reinnervation of the intermediolateral columns of the spinal cord at level C8–T2 by motor neurons of the extensor muscles of the hand occurred.
|
Views: 868
HTML: 468
PDF: 332
Figure 1: 0
Figure 2: 0
Table 1: 0
Authors affiliation: 0
Images captions: 0
Disclosure Author 1: 0
Disclosure Author 2: 0
Copyright Author 1: 0
Copyright Author 2: 0
|
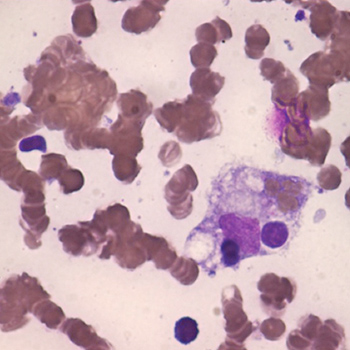
The authors describe a case of a 48-year-old man who presented with four weeks of fever, generalized malaise, weight loss, right upper quadrant abdominal pain and hepatosplenomegaly. He evolved with pancytopenia, bone marrow haemophagocytosis and hyperferritinaemia. Recent diagnosis of HIV infection, with the exclusion of other plausible causes, prompted the diagnosis of haemophagocytic syndrome (HPS) secondary to HIV. Despite intensive care support and initiation of antiretroviral therapy, the patient died. HPS diagnosis secondary to HIV alone demands the exclusion of all the other secondary causes. The best approach includes early diagnosis and specific treatment of the associated cause, whenever possible.
|
Views: 706
HTML: 282
PDF: 332
Fig 1: 0
Fig 2: 0
learning points: 0
|
Plasmodium infection in human beings is often associated with complications. Complications such as cerebral malaria, acute respiratory distress syndrome, acute kidney injury and cardiac complications including myocarditis, pericarditis and hypoglycaemia may be seen in infection by Plasmodium falciparum. However, these complications have rarely been reported with Plasmodium vivax infections. Myopericarditis complicating P. vivax malaria is particularly rare and only a few cases have been reported so far. We report on a case of myopericarditis due to P. vivax malaria to add to the literature
|
Views: 1838
HTML: 302
PDF: 330
Untitled: 0
Table and figure: 0
|
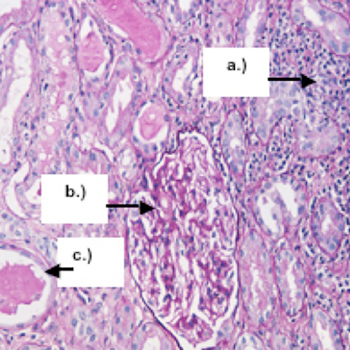
A 46-year-old female patient presenting with acute interstitial nephritis and anterior uveitis was admitted. The renal biopsy disclosed the presence of interstitial nephritis, confirming the clinical diagnosis of tubulointerstitial nephritis
and uveitis (TINU) syndrome. Treatment with oral steroids was started, with prompt improvement of symptoms and
laboratory abnormalities.
|
Views: 1621
HTML: 1016
PDF: 2087
|
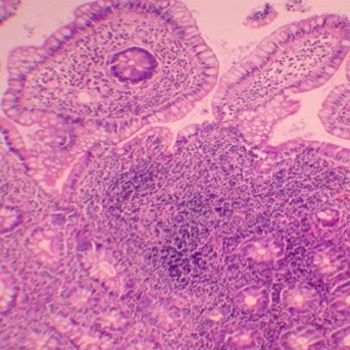
Crohn’s disease is a granulomatous in?ammatory bowel disease. Its pathologic ?ndings include non-contiguous chronic in?ammation and non-caseating granulomas, sometimes with extra-intestinal localizations. Sinonasal manifestations of Crohn’s disease are quite rare and only a few cases have been reported up to date in the worldwide literature. They are characterized by chronic mucosal in?ammation, obstruction, bleeding and occasionally septal perforation. We report a case of sinonasal granulomatosis revealing Crohn’s disease in a 22-year-old woman and go over the available literature on sinonasal involvement in Crohn’s disease.
|
Views: 1326
HTML: 610
PDF: 415
Untitled: 0
Untitled: 0
|
Introduction: Infective endocarditis (IE) has been reported to mimic granulomatosis with polyangiitis (GPA) and to test positive to antineutrophil cytoplasmic antibodies (ANCA), which may lead to a misdiagnosis and inappropriate treatment.
Case presentation: We report a case of a 59-year-old man admitted for purpura, gangrenous digital infarcts and glomerulonephritis. The diagnosis of IE was initially considered on the basis of heart murmur and two positive haemocultures to corynebacterium. Ineffectiveness of antimicrobial therapy and further neurological and nasal manifestations supported the diagnosis of GPA.
Conclusions: IE should be ruled out before initiation of immunosuppressive treatment. If the disease progresses despite antimicrobial treatment, vascular diseases should be rapidly taken into account in differential diagnosis and treated early to avoid fatal complications.
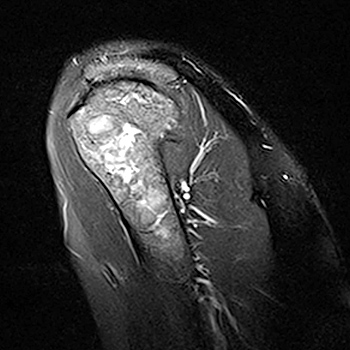
|
Views: 988
HTML: 575
PDF: 327
Figure 1: 0
Table 1: 0
|
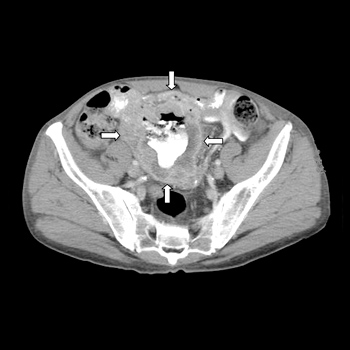
Objectives: Thrombocytopenia and splenomegaly are common features in several haematological disorders. Gaucher disease (GD) is a rare lysosomal storage disorder frequently characterized by thrombocytopenia and splenomegaly, which represents a clinical challenge for haematologists and internists.
Case: We describe the case of a 37-year-old patient with a diagnosis of spherocytosis since childhood, who developed hepatic failure and presented striking features of GD including hepatosplenomegaly, bone fractures and post-partum bleeding. We reconsidered the diagnosis of spherocytosis and investigated Gaucher disease.
Conclusion: GD should be considered in the differential diagnosis of thrombocytopenia and splenomegaly.
|
Views: 762
HTML: 246
PDF: 417
Covering Letter: 0
Author information page: 0
Fig. 1: 0
Fig. 2: 0
Fig. 3: 0
Copyright: 0
ADPCI1: 0
ADPCI2: 0
ADPCI3: 0
ADPCI4: 0
ADPCI5: 0
|
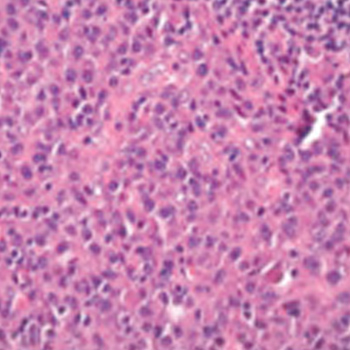
Objectives: We report a case of primary melanoma of the small intestine. Primary intestinal melanoma (PIM) is an extremely rare neoplasm for which the cause is unknown.
Materials and methods: A 67-year-old man was admitted to our department due to abdominal pain, constipation, a large, hard inguinal mass and severe anaemia.
Results: After laboratory data, imaging techniques and histopathological examination, the diagnosis was confirmed. A surgical resection of the intestinal neoplasm, treatment with BRAF inhibitors and radiation therapy to the inguinal mass were performed.
Conclusion: PIM is rare and it is usually difficult to establish its exact origin.
|
Views: 795
HTML: 1117
PDF: 515
|
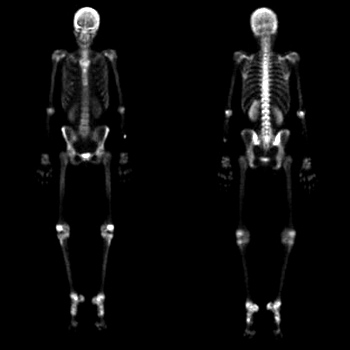
An 85-year-old male was hospitalized because of deterioration of his general condition and infection of the tracheostoma. He had had laryngectomy, bilateral neck dissection and radiation therapy for a laryngeal carcinoma 5 years earlier. Despite a good recovery, he could not get up because of a new onset of postural symptoms (dizziness, lightheadedness, collapse). Late onset of baroreflex failure and autonomic nervous system failure were diagnosed. Volatility of blood pressure (supine hypertension, upright hypotension) was treated with NaCl supplement during the day and a short-acting antihypertensive (clonidine) at night. With this regimen, the patient could walk without support.
|
Views: 1062
HTML: 902
PDF: 345
Reference Only: 0
Figure 1: 0
Figure 2: 0
Table 1: 0
Reference Only: 0
Reference Only: 0
|
A patient with Graves’ disease was admitted with a thyroid storm. She had severe hypercalcaemia caused by thyrotoxicosis. Treatment was complicated by vomiting and diarrhoea. With intravenous ondansetron, hydration and bisphosphonates, GI symptoms improved and oral thyreostatics could be started. This, combined with bisphosphonate administration, resulted in a mild hungry bone syndrome.

|
Views: 1074
HTML: 415
PDF: 408
Untitled: 0
Learning Points: 0
Revision as requested by reviewers: 0
|
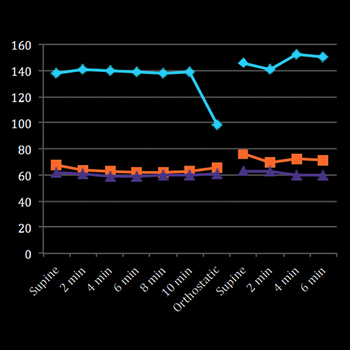
Objectives: To highlight the occurrence of Hashimoto’s encephalopathy – a steroid-responsive encephalopathy associated with elevated antithyroid antibodies.
Material and methods: We describe a clinically and biochemically euthyroid patient with an encephalopathy presenting with headache, mild confusion and personality changes for 6 weeks and tonic–clonic seizures upon admission
Results: There was no obvious infective or metabolic cause. The patient had a high titre of antithyroid antibodies and responded to steroid therapy.
Conclusion: This uncommon disease needs to be considered in patients presenting with neurological symptoms that remain unexplained after routine standard investigations, even when the patient is euthyroid. Early diagnosis is important, as this is a treatable condition.
|
Views: 924
HTML: 249
PDF: 326
Figure 1: 0
Table: 0
|
Lenalidomide is an effective therapy against malignant plasma cells and a potent agent against proinflammatory and proangiogenic cytokines. The use of lenalidomide in POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein with plasma cells, skin changes) has been reported, but its benefit in long-term use is not well established. A 55-year-old man with POEMS and debilitating polyneuropathy was treated with lenalidomide and dexamethasone followed by maintenance lenalidomide. He remains in haematologic remission and in complete recovery of functional status 3.5 years after diagnosis. This case supports the long-term use of lenalidomide in patients with POEMS syndrome.
|
Views: 1215
HTML: 1572
PDF: 362
cover letter: 0
consent form patient: 0
figure 1: 0
|
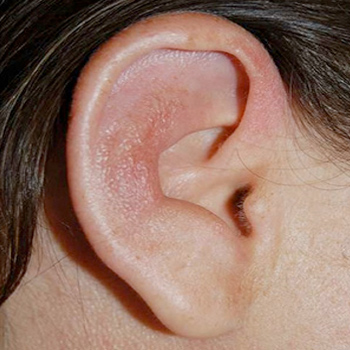
Introduction: Auricular chondritis has been occasionally described in patients with systemic lupus erythematosus (SLE) and antiphospholipid syndrome (APS).
Materials and methods: We report the case of a woman with a previous history of APS who presented with auricular chondritis with onset of SLE symptoms during the postpartum period.
Conclusion: SLE and APS should be taken into consideration in the differential diagnosis of auricular chondritis.
|
Views: 802
HTML: 260
PDF: 303
Figure 2: 0
Figure 1: 0
Figure 3: 0
Authors informations: 0
Covering Letter: 0
Signed copyrights forms and Disclosure Conflict of Interests forms: 0
Acknowledgment: 0
|
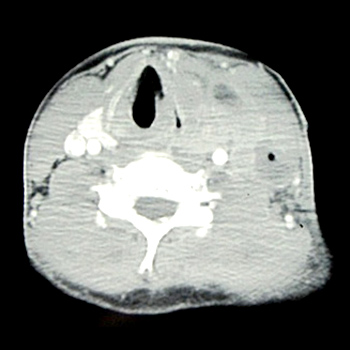
Objectives: To recall the common clinical presenting features of and guide the diagnostic procedures of primary thyroid lymphomas (PTL).
Materials and methods: We report on three patients developing an acute dyspnoea and fast evolving neck mass in the thyroid caused by a PTL occurring in our regional hospital in Switzerland between 2009 and 2013.
Results: PTL causes a neck mass, dyspnoea and dysphonia and responds well to chemotherapy. Mortality is due to relapse or infectious complications.
Conclusion: Acute dyspnoea caused by thyroid disease is uncommon. Nevertheless, it is the main symptom in PTL due to rapid growth and compression of the airways. Chemotherapy should be started promptly.
|
Views: 1152
HTML: 1007
PDF: 518
COVER LETTER: 0
|
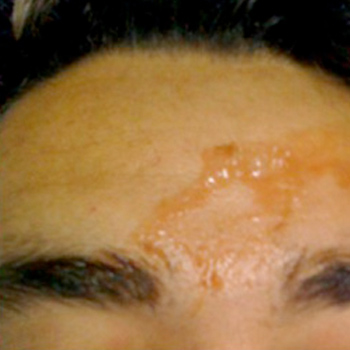
Objectives: Haematidrosis, also known as haematohidrosis, is a very rare condition where blood is excreted with sweat. As only a few cases have been described in the literature, we present guidelines on management of this rare phenomenon.
Case: A 44-year-old man presented with self-limited and spontaneous bleeding episodes from different parts of his body. This had started 2 weeks before admission after an episode of extreme emotional stress. Medical history and laboratory tests were normal. The microscopic examination of a sample of the fluid excreted confirmed all blood elements.
Conclusion: The disorder is thought to be related to activation of the sympathetic nervous system. The use of benzodiazepines and beta blockers may be helpful in controlling the bleeding episodes and give some comfort to the patient.
|
Views: 3572
HTML: 7319
PDF: 1474
Figure 2: Methemoglobin reduction:: 0
Figure 1: Methemoglobin trend over time: 0
Table 1: Arterial blood gas analysis on admission:: 0
|
Introduction: Various agents can lead to an acquired methaemoglobinaemia (MHB) with potentially fatal consequences. There is a lack of literature on the formation of methaemoglobin (MH) in the blood after the intake of poppers (amyl nitrite). Poppers are a popular aphrodisiac agent.
Case description: A 56-year-old diabetic called an ambulance after using poppers in a brothel with subsequent associated acrocyanosis, confusion and headache. The paramedics reported tachycardia and blood glucose of 3.8 mmol/l. The arterial blood gas analysis in the Emergency Department (ED) revealed a MHB of 23.1%. MH levels decreased rapidly without antidotal therapy. The patient was discharged the next day free of symptoms.
Discussion: This case illustrates the potential risks of taking poppers. A wide spectrum of symptoms were present in our patient. For the differential diagnosis of acquired MHB, poppers should be considered.
|
Views: 1073
HTML: 1044
PDF: 397
|
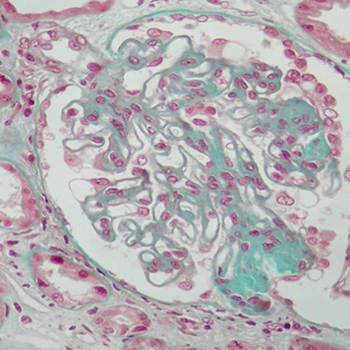
Introduction: Membranous glomerulonephritis is commonly described in systemic lupus erythematosus (SLE) and hypothyroidism.
Clinical presentation: We report a case of a 40-year-old woman who presented with a membranous glomerulonephritis associated with SLE, rheumatoid arthritis and hypothyroidism due to Hashimoto's thyroiditis.
Conclusions: The simultaneous occurrence of these three diseases as possible causes of membranous glomerulonephritis is extremely exceptional.
|
Views: 920
HTML: 358
PDF: 327
Echo: 0
The case with learning points: 0
Final revision: 0
|
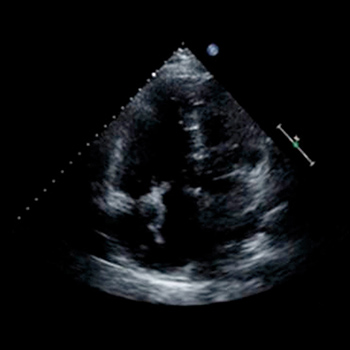
A 74-year-old man presented to our Emergency Department with acute dyspnoea. His electrocardiogram showed atrial flutter with 2:1 block and a rate of 150 bpm. Initial investigations revealed a D-dimer level of 6.01 mg/dl. Based on the patient’s complaints and the high D-dimer level, computed tomography pulmonary angiography was immediately performed. This showed no evidence of pulmonary embolism, but there were pneumatic changes in the right upper lung lobe. Antibiotics treatment was started with pipracillin/tazobactam, after which the patient’s condition improved. However, on the third day after admission he developed acute dyspnoea, diaphoresis and cardiopulmonary instability immediately after defecation. To promptly confirm our clinical suspicion of pulmonary embolism, a transthoracic echocardiography was carried out. This demonstrated a worm-like, mobile mass in the right heart. The right ventricle was enlarged, and paradoxical septal motion was present, indicating right ventricular pressure overload. The systolic tricuspid valvular gradient was 56 mmHg. The patient was treated with thrombolysis. His condition was greatly clinically improved after 3 hours. After 10 days of hospitalization, the patient was discharged.
|
Views: 1351
HTML: 1000
PDF: 535
Table 1: 0
Figure 1: 0
Cover letter: 0
|
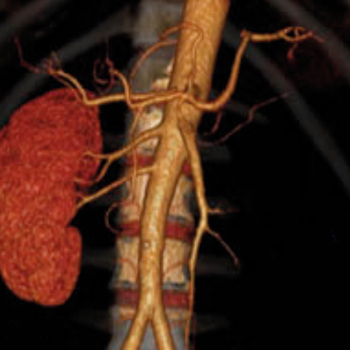
Renal artery thrombosis is a threatening clinical diagnosis, in which renal infarction may occur. Often misdiagnosed, for mimicking other common diseases, it should be considered in persistent flank pain to improve care and reduce morbidity. We review a case of a healthy, 57 year-old woman with renal artery thrombosis mimicking pyelonephritis and renal calculus obstruction, highlighting features of this clinical condition. An accurate diagnosis is essential for optimal management and prompts treatment, which still remains to be defined.

|
Views: 898
HTML: 772
PDF: 384
patient agreement: 0
|
Objectives: To contribute to current knowledge on the vascular risk of oestrogens.
Materials and methods: A 44-year-old woman received a 11.25 mg Leuprolide exteneded release injection to control bleeding from a 7 cm uterine fibroid tumour; 45 days later, she had a stroke due to right frontal lobe ischaemia. Thrombolysis induced complete remission. Three years previously, while taking a birth control pill, the patient had suffered from a stroke that involved her left temporal lobe. She was heterozygous for Factor V R2 H1299R locus and homozygous for the 4G/4G mutation of the PAI-1 gene. Even though her homocysteine level was normal, the patient was homozygous for the MTHFR C677T mutation and although she had never had severe bleeding, she was also homozygous for Factor XIII V34L.
Results and conclusion: This patient's prothrombotic condition could have been enhanced by leuprolide since its stimulatory effect on oestrogen production would still have been minimally present at the time of cerebral thrombosis.
|
Views: 1019
HTML: 856
PDF: 912
|
Autoimmune diseases may present as paraneoplastic syndrome. This is especially recognized in the case of polymyositis/dermatomyositis, but is less common in polymyalgia rheumatica. The authors describe the case of a 73-year-old man who presented with pain and stiffness of the scapular and pelvic girdles associated with asthenia lasting for a few weeks. The presence of therapeutic resistance and other atypical features directed the investigation towards the search of an occult malignancy. Patient evaluation revealed a pancreatic neuroendocrine tumour. After surgical treatment of the underlying neoplasia, the patient recovered fully with resolution of the rheumatic disease.
|
Views: 838
HTML: 540
PDF: 437
Table 1: 0
Table 2: 0
Figure 1: 0
figure 2: 0
|
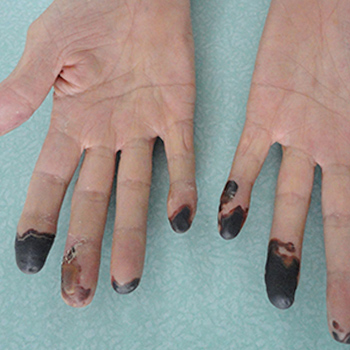
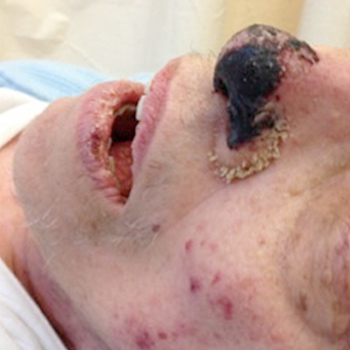
Multiple myeloma (MM) is a plasmocytic malignant proliferation of a single clone resulting in an overabundance of monoclonal immunoglobulins. MM commonly presents with bone disorders, renal failure, anaemia and hypercalcaemia. Hyperviscosity syndrome is rare, as are vaso-occlusive symptoms. The authors report a dramatic case of an 80-year-old woman admitted to the emergency department with full-blown distal gangrene. The culprit turned out to be a MM, unusually presenting with symptomatic hyperviscosity and peripheral occlusive ischaemia. This catastrophic and particularly dramatic presentation is almost unprecedented, with only a few cases reported worldwide.
|
Views: 1064
HTML: 1537
PDF: 387
Photo 1-2: 0
|
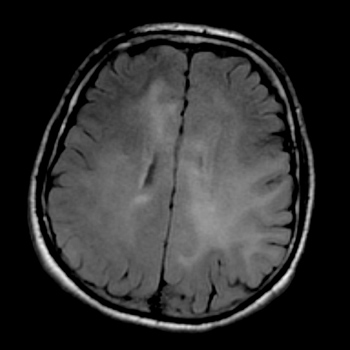
Hereditary angioedema (HAE) is a rare genetic disorder transmitted as an autosomal dominant trait, characterized by reduced plasma concentration or by the presence of non-functional C1 esterase inhibitor. Oedema caused by HAE mostly affects the skin and bowel and can induce swelling of genitalia. Oedema can be life threatening if it causes swelling of the larynx with obstruction of the airways.
We describe the case of a 52-year-old man who presented a neurological emergency (coma), where the remarkable localization of the clinical manifestation and the unusual symptomatology hindered the correct diagnosis.

|
Views: 2184
HTML: 1535
PDF: 417
copyright authorization form: 0
Sign Casco: 0
Sign Colmenero: 0
Sign Isasi: 0
Sign Fernandez Puga: 0
Sign Tejerina: 0
|
Introduction, objective: To present a case report in which the finding of non-coeliac gluten sensitivity was decisive for the treatment of a complex autoimmune disease.
Materials and methods: A 43-year-old woman with polyarthritis, psoriatic features, anti-SSA/Ro and anti-cyclic citrullinated peptide antibodies, with refractory course, was evaluated for gluten sensitivity despite negative serology for coeliac disease.
Results: The patient carried the HLA DQ2 haplotype and duodenal biopsy showed lymphocytic enteritis. A gluten-free diet resolved the clinical picture and permitted tapering of immunosuppressive therapy.
Conclusion: Non-coeliac gluten sensitivity can be associated with autoimmunity despite the absence of the specific autoantibodies of coeliac disease.
|
Views: 1554
HTML: 2863
PDF: 612
picture 1: 0
|
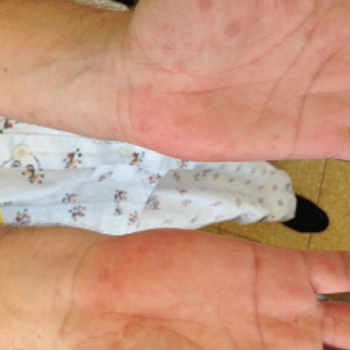
Syphilis is a sexually transmitted disease known to present with highly variable manifestations, especially when left untreated. Patients who present to Internal Medicine Departments with fever and a rash are always a diagnostic challenge since mild viral diseases and life-threatening bacterial infections may manifest themselves similarly. In the following case presentation, we describe a patient with 1 month’s duration of fever and rash on the palms of the hand and soles of the feet, in the form of erythema multiforme (EM)-like lesions. His disease was diagnosed as secondary syphilis, once again justifying its name: the “great masquerader".
| 2.1 = | 1.730 Cit. to date |
| 842 Docs. to date |




Publisher
Official Journal of the
European Federation of Internal Medicine
www.efim.org
Publisher: SMC media Srl
Via Giovenale, 7 - 20136 Milan - Italy
P.IVA 07626490960
info@ejcrim.com
www.ejcrim.com - ISSN: 2284-2594 - © EFIM 2014-2023, Published by SMC Media srl, Italy - Privacy policy

