ABSTRACT
This case report details the complex diagnostic odyssey of a 60-year-old female grappling with chronic liver disease, initially diagnosed with hepatic encephalopathy (HE). Despite initial treatment with lactulose and rifaximin, her neurological symptoms worsened, leading to the identification of concurrent acquired hepatocerebral degeneration (AHD). This condition is characterised by cognitive decline, movement disorders and distinctive imaging abnormalities. The discussion highlights the challenges in distinguishing AHD from HE, underscoring the sophisticated diagnostic and management strategies required for such intricate cases in the realm of chronic liver disease.
KEYWORDS
Chronic liver disease, hepatic encephalopathy, acquired hepatocerebral degeneration (AHD), liver transplant
LEARNING POINTS
- Recognizing coexisting conditions: emphasize the importance of identifying acquired hepatocerebral degeneration (AHD) alongside hepatic encephalopathy (HE) in patients with chronic liver disease. This recognition is crucial for comprehensive assessments and understanding the progression of neurological symptoms.
- Addressing management challenges: highlight the complexities of managing AHD due to limited therapeutic options and potentially irreversible outcomes. Discuss the challenges in decision-making, such as considering liver transplantation for patients with advanced neurological symptoms, and the need for exploring alternative therapeutic strategies.
- Conducting comprehensive evaluations: stress the significance of thorough evaluations in patients with chronic liver disease presenting with neurological symptoms. This comprehensive approach can help uncover underlying conditions like AHD, which may require different management strategies than those initially considered.
INTRODUCTION
Acquired hepatocerebral degeneration (AHD) is a rare disorder characterised by extrapyramidal symptoms, cognitive manifestations and movement disorders in individuals experiencing chronic liver disease (CLD) or portosystemic shunting[1]. It is noteworthy that AHD can either present independently or coexist with hepatic encephalopathy (HE), often being overlooked as a potential contributor to cognitive decline in liver disorders[2]. This case report discusses a patient whose hepatic encephalopathy symptom persisted despite comprehensive management, leading to the identification of AHD during an evaluation for liver transplantation. This highlights the importance of considering AHD in the differential diagnosis of neurological decline in the setting of CLD, especially when symptoms do not improve with standard treatment for HE.
CASE DESCRIPTION
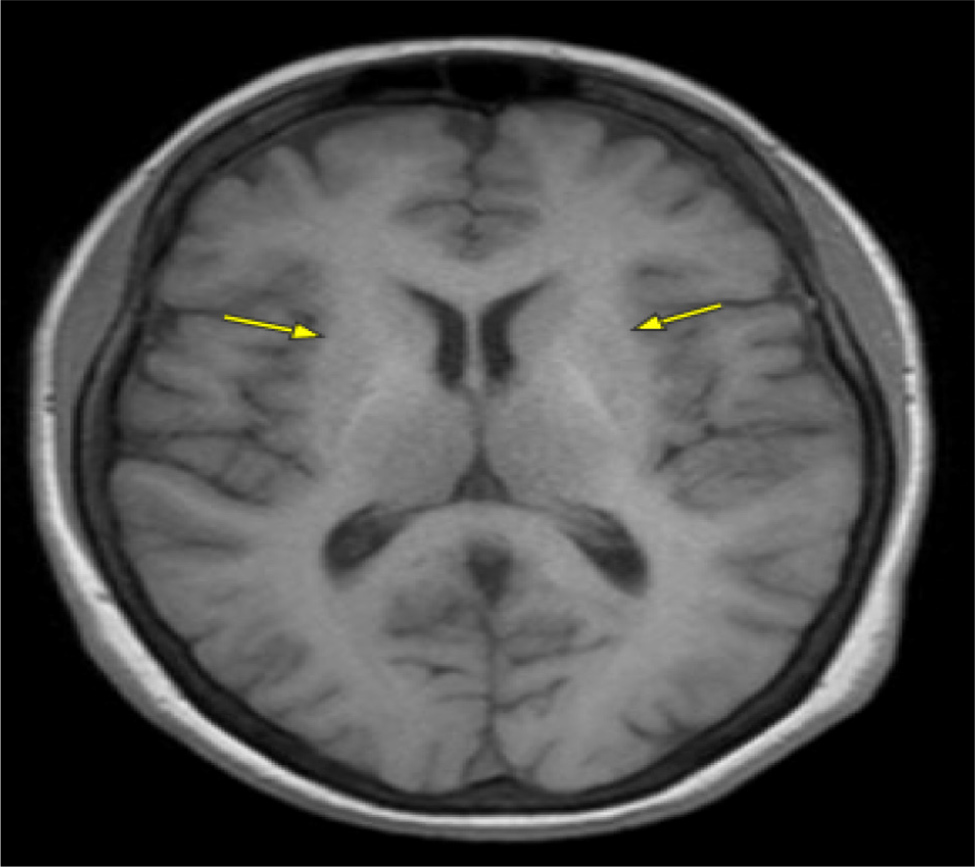
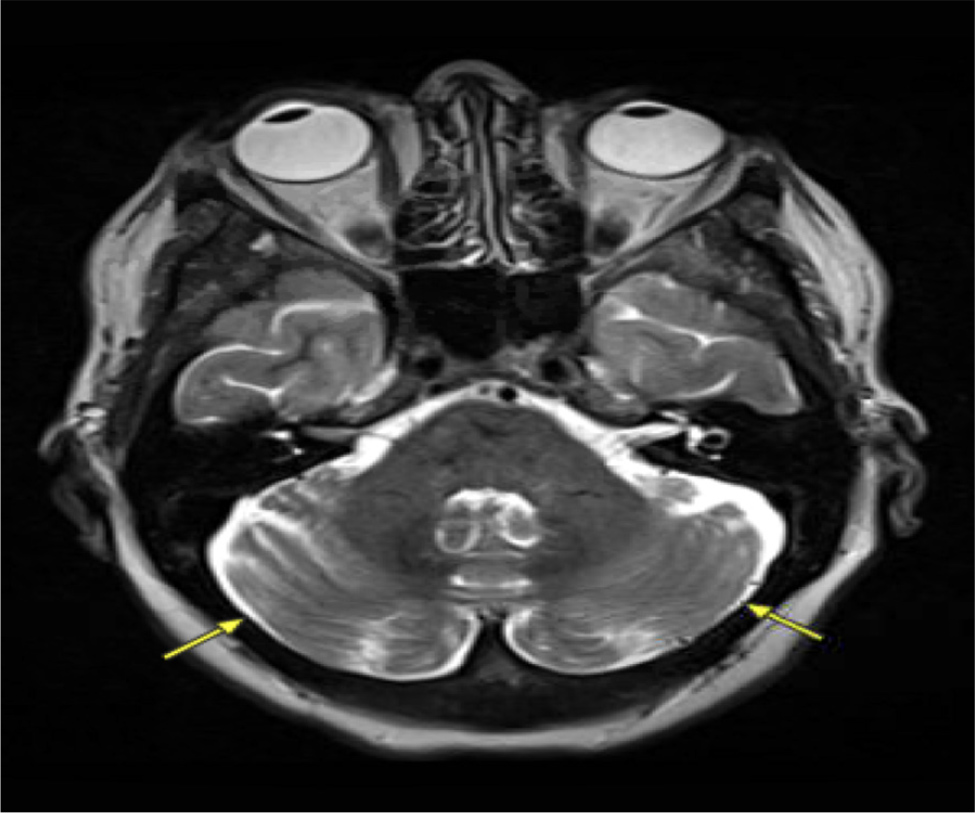
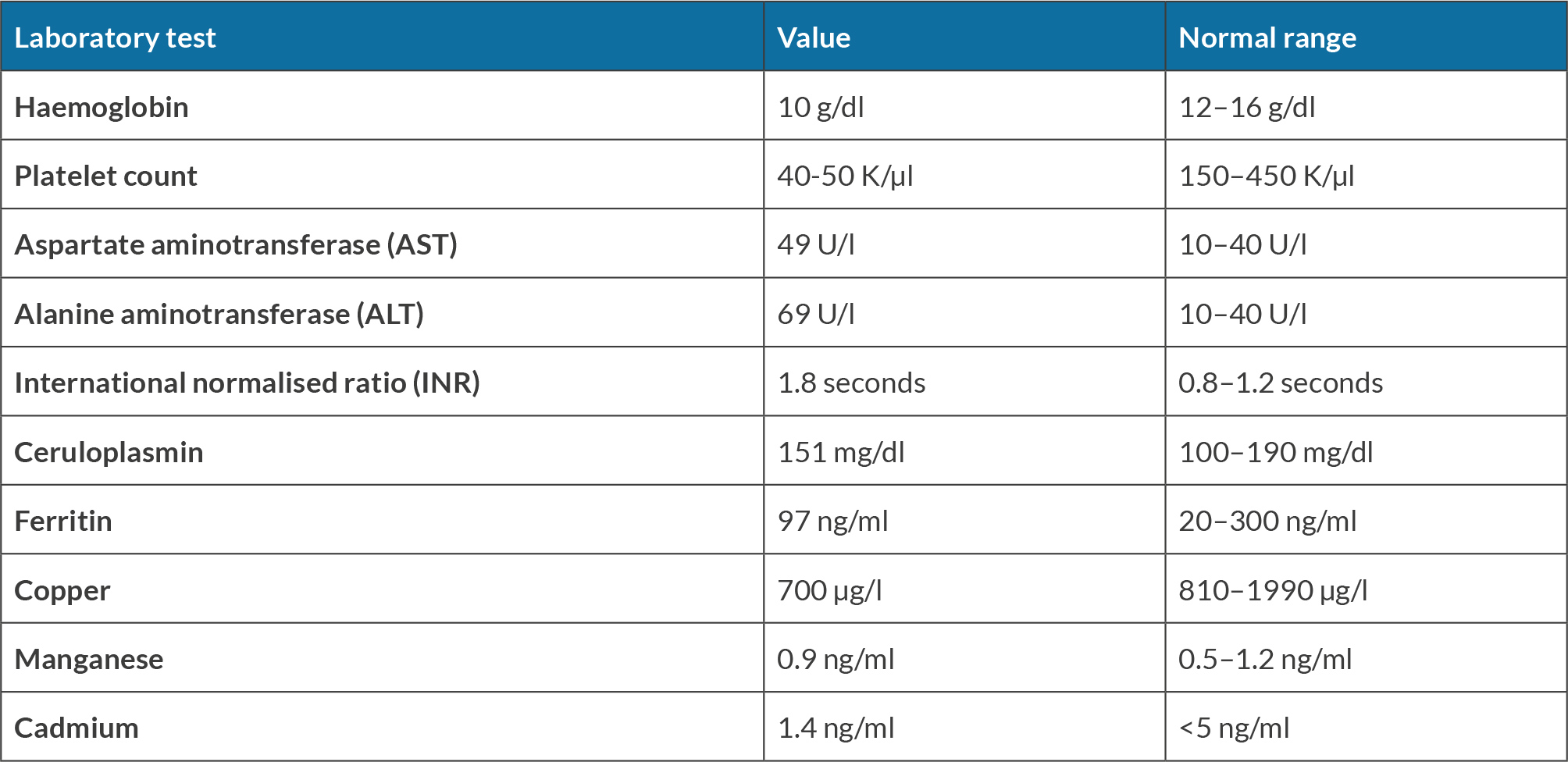
A 60-year-old female underwent liver transplantation evaluation at our centre due to decompensated cirrhosis with HE. The initial HE diagnosis occurred over two years ago following an emergency room visit for altered mentation, with computed tomography angiography (CTA) and magnetic resonance imaging (MRI) brain scans ruling out stroke. Subsequent abdominal imaging for hyperammonemia led to a new diagnosis of cirrhosis. The patient was treated with lactulose and rifaximin, which proved effective. However, over the previous year, the family noticed a progressive worsening of the patient’s mental status despite compliance with lactulose and rifaximin, necessitating multiple emergency room visits and culminating in a referral to the liver transplantation centre. At our centre, general neurology was initially consulted for increased falls and imbalance despite maximum lactulose and rifaximin doses. The neurology evaluation revealed difficulty comprehending new information, delayed processing and disorganised speech. Additionally, over the next six months, the patient was diagnosed with tardive dyskinesia without prior neuroleptic exposure, and electromyography (EMG) showed sensory neuropathy. The movement disorders team recognised sensory gait ataxia due to polyneuropathy with mild involuntary movements of the mouth. Various laboratory assessments on different dates highlighted significant findings. The complete blood count revealed consistently low levels of haemoglobin, averaging 10 g/dl (normal range: 12–16 g/dl), along with persistently low platelet counts ranging around 40–50 K/µl (normal range: 150–450 K/µl). Liver tests revealed borderline elevated levels, with aspartate aminotransferase at 49 U/l (normal range: 10–40 U/l) and alanine aminotransferase at 69 U/l (normal range: 10–40 U/l), alongside an international normalised ratio (INR) of 1.8 seconds (normal range: 0.8–1.2 seconds). A liver biopsy confirmed metabolic-associated steatohepatitis (MASH) with cirrhosis. The model for end-stage liver disease MELD-Na score was 14 upon the initial presentation, increasing to 23 over two years. The CLD workup showed negative viral/HIV markers, along with normal blood ceruloplasmin (20 mg/dl, normal range: 15–45 mg/dl), normal alpha-1 antitrypsin levels (151 mg/dl, normal range: 100–190 mg/dl), and normal ferritin levels (97 ng/ml, normal range: 20–300 ng/ml). MRI brain scans from two years previously during the first emergency room visit were reviewed and confirmed to have no acute intracranial abnormalities. A subsequent evaluation by behavioural neurology for progressive cognitive decline indicated sensory ataxia, neuropathy, dysarthria and executive dysfunction in the context of HE due to decompensated cirrhosis. Brain imaging revealed a mild hyperintense putaminal ring with no evidence of multiple system atrophy, but with mild parkinsonian features suggesting neurodegeneration. Follow-up MRI by neurology highlighted hyperintensities in the globus pallidus and subthalamic region on T1 (Fig. 1), with increased T2/FLAIR signal in the middle cerebellar peduncles and putamen, indicative of cerebellar degeneration (Fig. 2). Toxin and heavy metal screening indicated mildly decreased copper levels (700 µg/l, normal range: 810–1990 µg/l), alongside normal manganese levels at 0.9 ng/ml (normal range: 0.5–1.2 ng/ml) and cadmium levels at 1.4 ng/ml, falling within the normal range (<5.0 ng/ml). All the pertinent laboratory results are shown in Table 1. A skin biopsy for neurodegenerative protein biomarkers was negative for alpha-synuclein pathology e.g. Lewy body disease, confirming a diagnosis of AHD.
(click to enlarge)
Figure 1. Hyperintensities in the globus pallidus (yellow arrow), subthalamic region and midbrain on T1.
(click to enlarge)
Figure 2. MRI brain showing an increased T2/FLAIR signal in the middle cerebellar peduncles, raising concerns for degeneration.
(click to enlarge)
Table 1. Patient laboratory test results and normal ranges.
The patient transitioned from not using any mobility aid to using a walker over six months. The progressively worsening cognitive symptoms despite HE therapy, alongside progressive motor function decline with imaging suggestive of injury to the globus pallidus, were consistent with a diagnosis of AHD. As per recommendations, two cycles of manganese chelation therapy were attempted with minimal to no improvement. However, given the deteriorating neurological status, the decision was made to decline liver transplantation, leading to a transition to hospice care.
DISCUSSION
Conditions affecting the liver can give rise to diverse neurological impairments, with HE being the most prevalent[3]. AHD is a rare neurological condition observed in patients with CLD or portosystemic shunts[3]. It is characterised by manifestations such as parkinsonism, cognitive decline and movement disorders, forming a distinct profile of neurological complications[1]. The diagnosis of AHD relies on specific clinical criteria, including the presence of neurological manifestations, hyperintensity of the globus pallidus observed on T1-weighted brain MRI and the concurrent occurrence of CLD[4].
Our patient exhibited all manifestations of AHD gradually after the diagnosis of HE. Initial overlapping cognitive symptoms were treated for recurrent symptoms of HE with some improvement, posing a challenge in early diagnosis of AHD in our case. It is important to note that AHD and HE can coexist, as seen in the case mentioned by Huang[5] and the research study involving 76 patients conducted by Malaquias[6]. Huang’s case involved a presentation with extrapyramidal symptoms and coma. The classic findings on the MRI during admission led to the diagnosis of AHD.
The Malaquias study highlighted that AHD often coexists with HE, as evidenced by their simultaneous diagnosis in 82.9% of 76 individuals studied[6]. Parkinsonism syndrome was found in 44.7% of patients, with attention and initiation/perseveration being the most affected cognitive areas, similar to our patient[6]. Of note, this is the first case report to the knowledge of the authors that has confirmed the absence of alpha-synuclein disease in AHD using skin protein biomarkers. During the follow-up period, 42.7% of patients passed away, and 25% underwent liver transplantation, highlighting the serious consequences and challenges associated with this condition. Interestingly, patients in the AHD group exhibited longer survival (3 deaths) compared to the AHD with HE group (29 deaths), although no significant differences were observed regarding the coexistence of HE[6]. This highlights a noteworthy aspect – AHD patients with co-existent HE may face a potentially more unfavourable prognosis.
The development of AHD may be linked to the accumulation of toxic substances such as manganese or ammonia, which is facilitated by compromised liver clearance. Increased ammonia levels in CLD impede glutamate removal, resulting in glutamine accumulation and mild brain oedema[7]. This heightened ammonia concentration can activate nitric oxide synthase, contributing to nitrosative stress in the central nervous system. These effects may synergise with pro-inflammatory cytokines and toxic metals, such as manganese[7]. Nonetheless, the manganese theory has gained more support over the last twenty years, with several studies highlighting it as a key player in the pathogenesis of AHD[8]. The hepatobiliary system is responsible for clearing manganese from both the blood and the cerebrospinal fluid. However, in certain patients manganese concentrations exceed expected levels due to toxic substances evading clearance by the hepatobiliary system and entering systemic circulation[7]. This anomaly may be attributed to portosystemic shunts and liver dysfunction. Consequently, the brain becomes susceptible to manganese deposition, primarily in areas such as the basal ganglia, brainstem, cerebral cortex and surrounding white matter. Importantly, it is crucial to recognise that manganese levels are not consistently elevated in AHD patients, as seen in our patient. Malaquias’s comprehensive study also addressed the diagnostic marker of hypermagnesemia, indicating that only 38% of patients exhibited elevated manganese levels, highlighting its weak diagnostic significance in AHD[6].
In the presented case, evidence of liver cirrhosis and a liver biopsy confirming MASH is noteworthy. The potential presence of undetectable shunts on ultrasonography, especially in cases of micro shunts, should be considered[9]. Commonly observed magnetic resonance features encompass T1 hyperintensity in the substantia nigra, pallidum, periaqueductal grey matter, and occasionally, the dentate nucleus[9]. This heightened signal is currently attributed to manganese deposition, supported by postmortem studies indicating levels up to seven times the normal range in the globus pallidus. Our patient’s MRI performed two years previously for stroke evaluation did not show any cranial abnormality. It had progressed, and findings were consistent with those typical of AHD[10].
Therapeutic interventions for chronic AHD remain sparse and for most patients, the condition proves to be irreversible[11]. There have been reports suggesting that certain patients with AHD show positive responses to levodopa therapy or rifaximin. For a select group of patients, liver transplantation has demonstrated efficacy in treating AHD[8]. Unfortunately, the advanced neurological symptoms commonly presented by many patients often result in their exclusion from the transplantation waiting list, which happened with our patient. This exclusion underscores the pressing need for alternative therapeutic approaches and early diagnosis for managing this debilitating condition.