ABSTRACT
Background: We describe a case of myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) in a 16-year-old patient who initially presented with clinical features of septic meningoencephalitis. This case outlines the importance of considering a diagnosis of MOGAD in patients who fail to improve with appropriate antimicrobial therapy or show a positive clinical response to glucocorticoids (often used in treatment of meningococcal meningitis). We emphasise the importance of recognising that an infectious prodrome can precede MOGAD.
Case description: A 16-year-old male was admitted with vomiting, fever, headache, photophobia and altered mental state. He was treated for meningoencephalitis with initial clinical improvement. Lumbar puncture findings were suggestive of viral meningoencephalitis. During admission the patient went through several periods of transient clinical and biochemical improvement, alternating with periods of symptomatic relapse. On day 17 of admission, he was transferred to a tertiary centre for suspected autoimmune disseminated meningoencephalitis (ADEM) and two days later, he suffered a catastrophic neurological decline with new dysarthria, dysphagia, aphasia, horizontal nystagmus and facial paralysis. He made a remarkable neurological recovery after commencing treatment with IV immunoglobulin, IV methylprednisolone and plasma exchange, with complete resolution of symptoms.
Conclusion: MOGAD can run a variable course and present soon after a central nervous system infection, making the diagnosis more challenging. Nonetheless, patients can achieve a full neurological recovery with early recognition, diagnosis and treatment of this rare entity.
KEYWORDS
MOG antibody, meningoencephalitis, acute disseminated encephalomyelitis
LEARNING POINTS
- Autoimmune encephalitis can be preceded by an infectious prodrome which makes the diagnosis more challenging.
- Autoimmune encephalitis can run a subacute and fluctuating course with transient periods of symptomatic improvement preceding a rapid neurological decline.
- Glucocorticoids often used in treatment of patients with meningococcal meningitis may lead to transient symptomatic improvement in patients with autoimmune encephalitis, masking the diagnosis.
- MRI findings of demyelination in autoimmune encephalitis may lag behind clinical symptoms by days to weeks.
INTRODUCTION
We describe a case of myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) in a 16-year-old patient who presented with clinical features of septic meningoencephalitis. This case outlines the importance of considering a diagnosis of MOGAD in patients who fail to improve with appropriate antimicrobial therapy or show a positive clinical response to glucocorticoids (often used in treatment of meningococcal meningitis). We emphasise the importance of recognising that an infectious prodrome can precede MOGAD and MRI findings of demyelination may lag behind clinical symptoms.
CASE DESCRIPTION
A 16-year-old male presented with fever, meningism and psychosis. He was normally well and did not take any regular medications. The initial blood tests revealed an elevated white cell count of 26 x 109/l (normal range 4.5–13 x 109/l), neutrophil count of 24.05 x 109/l (normal range 1.8–8.0 x 109/l), low lymphocyte count of 1.13 x 109/l (normal range 1.2–5.2 x 109/l), and C-reactive protein (CRP) of 66 mg/l (normal range 0–5 mg/l).
The COVID-19 polymerase chain reaction (PCR) was negative. The international normalised ratio was 1.5 (normal 2–4.5), and prothrombin time was 18.1 seconds (normal 10.7–13.9 seconds). Sepsis-driven coagulopathy was suspected, and 10 mg vitamin K was given to correct this. The CT and MRI scans of the patient’s brain were unremarkable. The clinical impression was meningoencephalitis, for which he was given IV ceftriaxone 2 g twice daily, acyclovir 500 mg IV four times daily and dexamethasone 10 mg IV immediately.
After 48 hours, the patient reported an improvement in meningism, and was apyrexial. The cerebrospinal fluid (CSF) analysis revealed an elevated white cell count with pleocytosis of 86 mm3 (20% polymorphs, and 80% lymphocytes); normal total CSF protein of 0.4 g/l (normal range 0.15–0.45 g/l); clear and colourless appearance; normal CSF glucose of 3.1 mmol/l (normal range 2.2–3.9 mmol/l). Microscopy, culture and sensitivity showed no organisms on Gram film with no bacterial growth, and viral PCR was negative. He remained emotionally labile until day 5 of admission. The CRP fell to 10.7 mg/l, white cell count was 11.30 x 109 and neutrophils were 9.16 x 109/l. He was treated for a viral meningoencephalitis. Viral PCR was considered to be negative due to early antimicrobial treatment; ceftriaxone was discontinued on day 4. Some 24 hours after stopping IV ceftriaxone, the patient became febrile (39°C) and reported a recurrence of headache and meningism; he developed urinary retention (643 ml) and was catheterised. He developed brisk lower limb reflexes and upgoing plantars bilaterally. This clinical deterioration was mirrored by a rise in inflammatory markers: the CRP rose to 42.5 mg/l, white cell count to 19.4 x 109/l and neutrophils to 16.33 x 109/l; IV ceftriaxone was restarted. A repeat contrast CT head was normal. IV dexamethasone was restarted due to clinical features of suspected cord compression and 24 hours after restarting dexamethasone and ceftriaxone, the patient reported resolution of headaches and meningism, and was apyrexial. His inflammatory markers improved, and a repeat MRI head and spine was normal. Therefore, the dexamethasone was stopped (after receiving a total of five doses, 10 mg each) and the patient remained well for the next five days, on IV ceftriaxone and acyclovir.
Five days later, he suffered a recurrence of severe headaches and became febrile (38°C). He had a second lumbar puncture which showed elevated white cells with pleocytosis of 196 mm3 (90% lymphocytes, 10% polymorphs); viral PCR was negative, 16S PCR was negative, glucose was 2.2 mmol/l, protein 0.8 g/l (normal range 0.15–0.45 g/l), clear and colourless, with an elevated opening pressure of 27 mm CSF. Extended viral, Lyme disease, hepatitis and retroviral serology were negative. An autoimmune and vasculitis screen was negative. Ongoing meningoencephalitis of infective aetiology was suspected, and an autoimmune process was considered to be unlikely given the normal MRI scans and clinical improvement on IV ceftriaxone.
On day 17 of admission, the patient became delirious and agitated, with a recurrence of urinary retention (900 ml). Due to a failure to respond to treatment for infective meningoencephalitis, a plan was made to transfer him to a tertiary centre, for management of suspected autoimmune disseminated meningoencephalitis (ADEM). Two days later, the patient suffered a catastrophic neurological decline, with new dysarthria, dysphagia, aphasia, horizontal nystagmus and facial paralysis. In the lower and upper limbs, he developed increased tone, brisk reflexes and 0/5 power. He then developed bradycardia, hypertension and airway compromise, which suggested brainstem involvement. He was intubated and ventilated, and treated with 1 g IV methylprednisolone for 5 days, IV immunoglobulin and double plasma exchange. After this, he was started on 40 mg of prednisolone. A repeat MRI with contrast showed typical ADEM changes, with widespread neuroinflammatory changes and T2 weight hyperintensities in the juxtacortical white matter, bilateral thalami, pons, corticospinal tracts and cerebellum (Fig. 1-3). There was abnormal signal change in the left optic nerve and longitudinally extensive transverse myelitis spanning almost the entire length of the spinal cord (Fig. 4). The anti-myelin oligodendrocyte glycoprotein (MOG) antibodies returned positive in high titres; the aquaporin-4 antibodies were negative. The clinical impression was therefore MOG antibody disease (MOGAD)-mediated ADEM. After two weeks on the neurorehabilitation ward, the patient made a dramatic improvement and was able to mobilise with a frame. There were no further difficulties with his mental state, speech or swallowing. A slow steroid wean with prednisolone was continued after discharge. He eventually returned to school and has had no further relapse during follow-ups for the last 18 months.
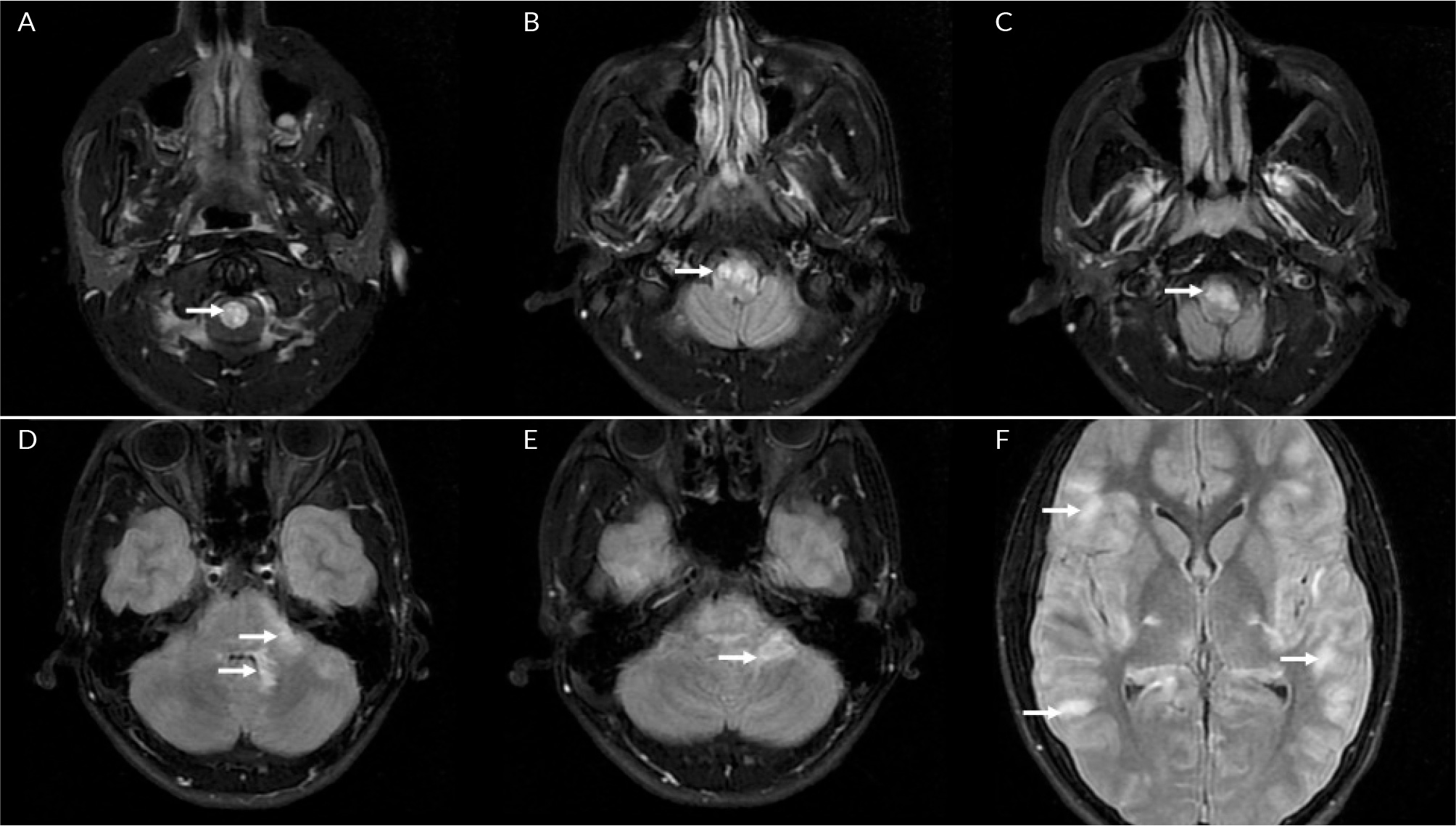
(click to enlarge)
Figure 1. T2/FLAIR weighted axial MRI brain images (lesions indicated with arrows): A) T2 signal abnormality in the corticospinal tracts at the level of the cervical spinal cord; B, C) T2 signal abnormality at the level of the medulla; D, E) T2 signal abnormality with preferential involvement of the peripheral pons, the adjacent left middle cerebellar peduncle and both superior cerebellar peduncles and the ventral medulla; F) Diffuse FLAIR hyperintensity within the convexity subarachnoid spaces.
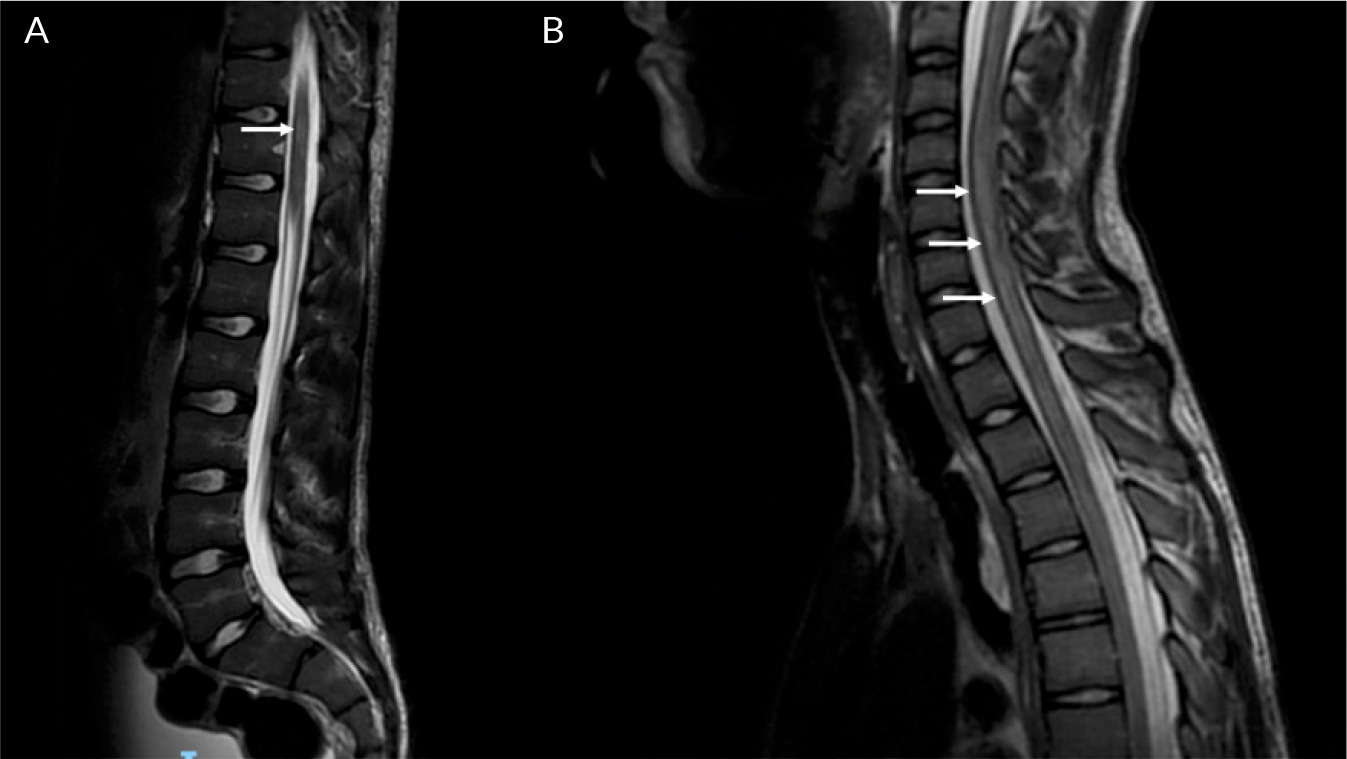
(click to enlarge)
Figure 2. T2 weighted sagittal MRI spine images (lesions indicated with arrows): A) T2 signal hyperintensity in the thoracic spinal cord; B) diffuse T2 signal abnormality in the cervical spinal cord.
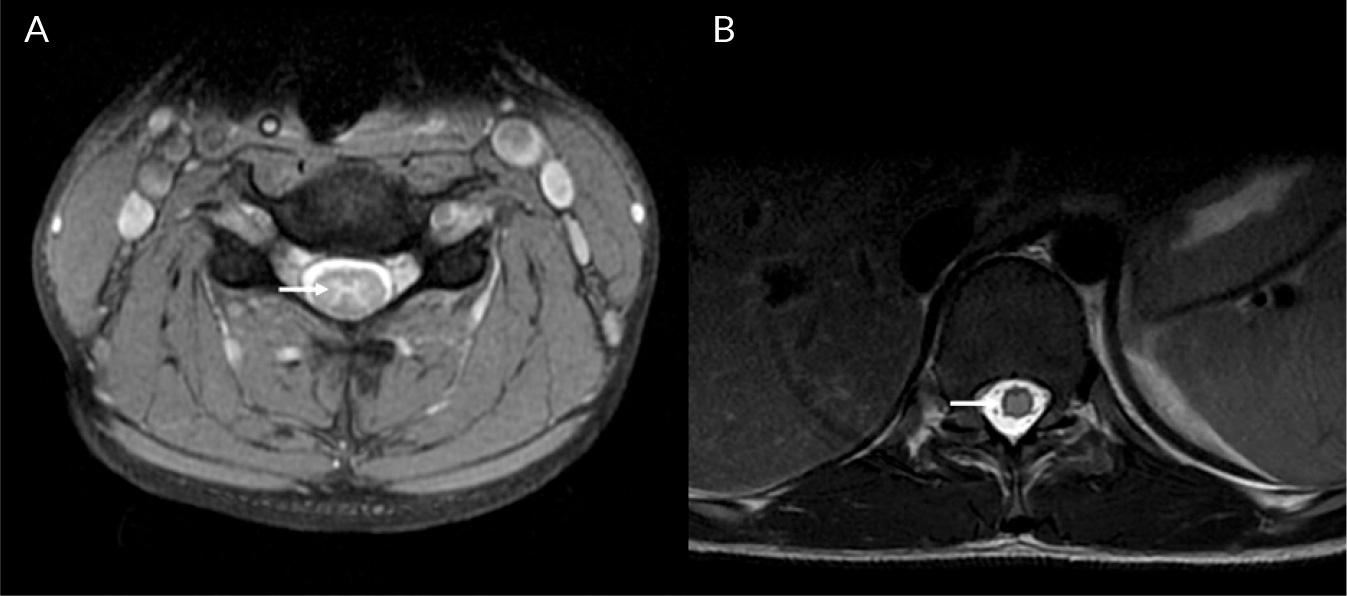
(click to enlarge)
Figure 3. T2 weighted axial MRI spine images (lesions indicated with arrows): A) butterfly shaped T2 signal hyperintensity in the cervical spinal cord; B) diffuse T2 signal abnormality in the thoracic spinal cord.
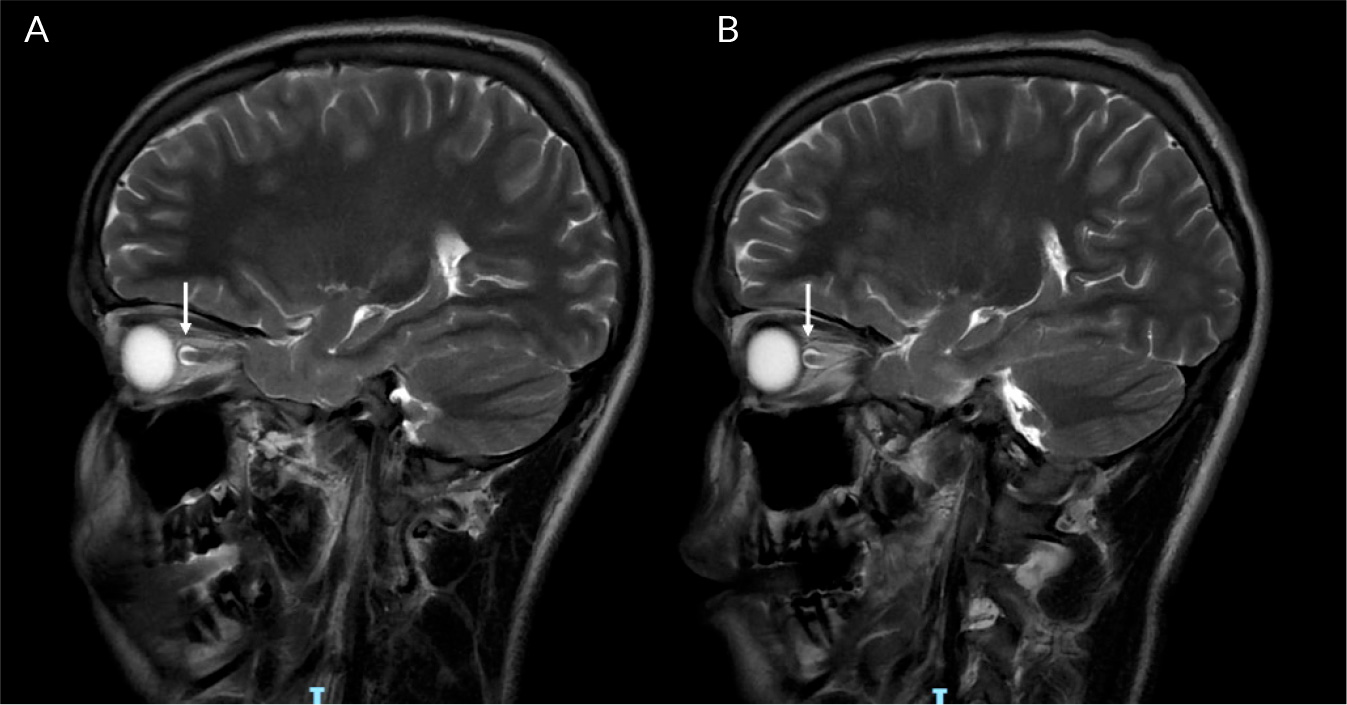
(click to enlarge)
Figure 4. T2 weighted sagittal MRI brain images (lesions indicated with arrows): A) T2 signal hyperintensity in the right optic nerve indicating optic nerve swelling; B) T2 signal abnormality in the left optic nerve indicating optic nerve swelling.
DISCUSSION
ADEM is an acute inflammatory demyelinating disease of the central nervous system (CNS). ADEM has been described in the literature since the eighteenth century, although the precise pathophysiology is still not fully understood[1]. It primarily affects the paediatric population, with a preceding infectious prodrome in 70–80% of cases[2,3]. A diagnosis of ADEM requires the presence of encephalopathy not attributed by fever, polyfocal CNS symptoms, MRI findings of diffuse, poorly demarcated inflammatory lesions predominantly involving cerebral white matter. It shows no new symptoms, signs or MRI findings after three months of the index presentation. Progression is usually subacute and takes place over several days.
Recent studies have described an association of the myelin oligodendrocyte glycoprotein antibody (MOG-Ab) with ADEM. MOGAD is a spectrum of clinical syndromes including ADEM, transverse myelitis, optic neuritis, aseptic meningitis and meningoencephalitis. In the paediatric population, ADEM is the most common type of MOGAD. The demographic and clinical characteristics are mostly indistinguishable between ADEM patients who test positive for MOG-Ab, compared to those who test negative, with a few exceptions. Patients with MOG antibody-associated ADEM are more likely to have spinal cord involvement and fewer emotional symptoms, compared to ADEM patients who tested negative for the MOG-Ab (93% vs 33% and 5% vs 43%, respectively)[4]. Patients with MOG antibody-associated ADEM have a higher number of white blood cells in CSF than MOG-Ab negative ADEM patients.
The prevalence of MOGAD is 1.3–2.5/100,000, and the annual incidence is 3.4–4.8 per million[5]. International diagnostic criteria for MOGAD require the presence of at least one of the core clinical demyelinating events as described above, in addition to a positive MOG-IgG test, and the exclusion of alternative diagnoses including multiple sclerosis[6]. Standard treatment for MOGAD and ADEM commonly includes IV steroids, plasmapheresis and intravenous immunoglobulin. Long-term treatments include oral steroids, with occasional off-licence use of IL-6 receptor blockers (e.g. tocilizumab).
In this case, the patient initially presented with meningoencephalitis of likely infectious aetiology. The CSF analysis revealed marked pleocytosis, which may be consistent with a viral aetiology although it is also typical of MOGAD[7,8]. The patient showed clinical and biochemical improvements on antimicrobial therapy and acute deterioration when it was stopped. This raised suspicions of bacterial meningoencephalitis. However, this pattern could be confounded by brief periods of IV dexamethasone therapy given twice during his hospital stay, with sustained clinical response likely attributed to its long biological half-life of 36–54 hours. Early on in his admission, the patient did not meet the radiological diagnostic criteria for MOGAD or ADEM. An alternative explanation for his condition was available, i.e. infectious meningoencephalitis, which is an exclusion criterion for MOGAD. The lack of identified microorganism in the CSF is a limitation of this case study, attributed to early initiation of antimicrobial therapy.
Later in his admission, the patient suffered severe headaches and urinary retention with upper motor neurological signs in the lower limbs. It is likely that at this point, he developed MOG antibody-associated transverse myelitis. He promptly received five doses of IV dexamethasone 10 mg, which was stopped when the MRI spine result returned unremarkable. He quickly improved clinically and regained his bladder function. Because the MRI spine was performed only 24 hours after the onset of symptoms of spinal cord involvement, it may have been too early to detect demyelination. Literature suggests that MRI findings of demyelination may lag behind clinical symptoms by days to weeks[9,10]. It is very likely that the patient then went on to develop MOG antibody-associated ADEM five days later, when he deteriorated clinically with recurrence of encephalopathy and urinary retention. This may correspond to the reduction in the pharmacological activity of IV steroids. Eventually, the MRI findings in the patient became apparent several days after a catastrophic clinical decline with brainstem involvement.