ABSTRACT
Background: Hypertrophic pachymeningitis (HP) is a disease with diverse aetiologies, including the autoimmune one, either associated with antineutrophil cytoplasmic antibodies or immunoglobulin G4.
Case description: A 65-year-old woman with a history of systemic arterial hypertension, presented with intense progressive headaches. HP and hemispheric vasogenic oedema were observed by nuclear magnetic resonance (NMR) study. During the six months before the headache, she had developed progressive hearing loss which she attributed to age. A biopsy of dura mater showed necrotising vasculitis with peripheral inflammatory infiltrate, made up of accumulations of epithelioid cells and multinucleated giant cells, and abundant eosinophils. A final diagnosis of HP with eosinophilic granulomatosis with polyangiitis (EGPA) was made.
Discussion: The patient had eosinophilic granulomatosis with polyangiitis (EGPA) histology, ANCA-negative serology and HP. This case is important because it shows that EGPA seems to have a spectrum of clinical diseases, including HP with negative serology, and bilateral sensorineural hearing loss.
Conclusion: We are facing a wide spectrum of EGPA, breaking the paradigm of only systemic involvement.
KEYWORDS
Headache, bilateral sensorineural hearing loss, hypertrophic pachymeningitis, eosinophilic granulomatosis with polyangiitis
LEARNING POINTS
- Hypertrophic pachymeningitis (HP) has several aetiologies; if the systemic investigation is not contributory to a diagnosis, a meningeal biopsy is necessary.
- This is the first case report of HP, associated with eosinophilic granulomatosis with polyangiitis (EGPA), and ANCA-negative serology.
- EGPA is probably a spectrum of diseases with predominant systemic involvement, but there may be cases where there is histological evidence, without the systemic context or positive serology.
INTRODUCTION
Hypertrophic pachymeningitis (HP) is the thickening of the dura mater secondary to an inflammatory process with diverse aetiologies. Patients have a headache and involvement of cranial nerves, but can be asymptomatic[1]. We present a case of HP, associated with eosinophilic granulomatosis with polyangiitis (EGPA), and negative antineutrophil cytoplasmic antibodies, limited to the intracranial compartment.
CASE DESCRIPTION
A 65-year-old woman with controlled arterial hypertension began with a left hemicranial headache of moderate to severe intensity, throbbing, exacerbated by movement, with occasional extension to the entire head. The headache improved with non-steroidal analgesics, but improvement decreased over the course of one year. Two weeks before admission, she developed weakness and a sensation of stiffness in the lower extremities that prevented her from walking for 24 hours, with apparently complete spontaneous remission. Finally, she went to the hospital for evaluation during which a symmetrical bilateral hearing loss and bilateral palmomental reflex were detected.
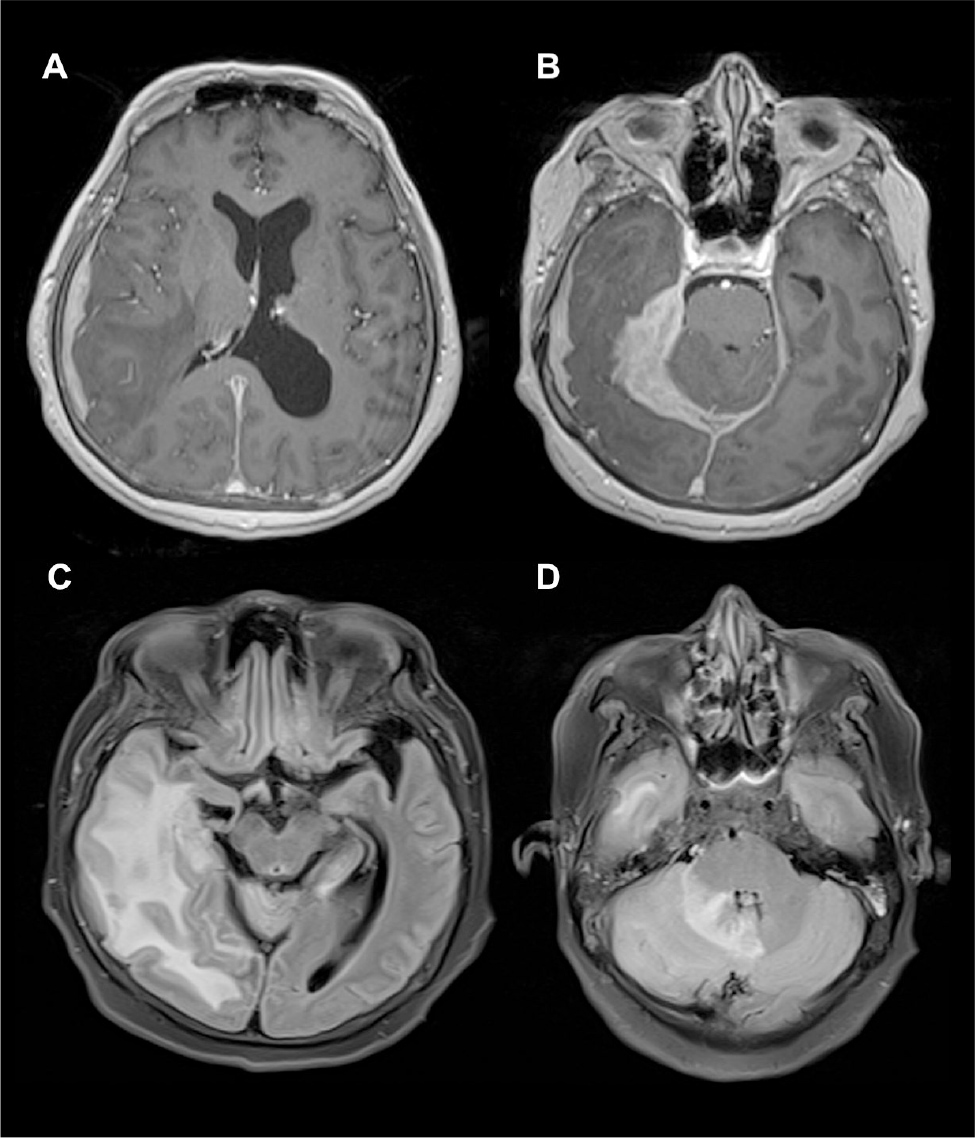
Upon guided questioning, the patient reported progressive bilateral hearing loss that started six months before the headache, which she attributed to age. The initial magnetic resonance imaging (MRI) study of the brain, after the administration of contrast medium, showed the following in the axial slices of the T1-weighted images: focal enhancement and irregular thickening of the pachymeninges at the temporal level with dorsal and ventral extension, and diffuse and irregular thickening and strong enhancement of the tentorium. In addition, T2-weighted FLAIR images revealed an extensive subcortical oedema in the right temporal lobe, with obliteration of the ventricular temporal recess, and oedema of the right cerebellar hemisphere that was extended and involved the vermis (Fig. 1).
(click to enlarge)
Figure 1. RMN. Axial slices of T1-weighted images with gadolinium showed pachymeningitis in the temporal region (A), with dorsal and ventral extension (B). Diffuse and irregular thickening of the tentorium on the right side is noted, with strong enhancement to contrast medium (B). T2-weighted FLAIR images showed subcortical oedema in the right temporal lobe (C), and in the right cerebellar hemisphere and vermis (D).
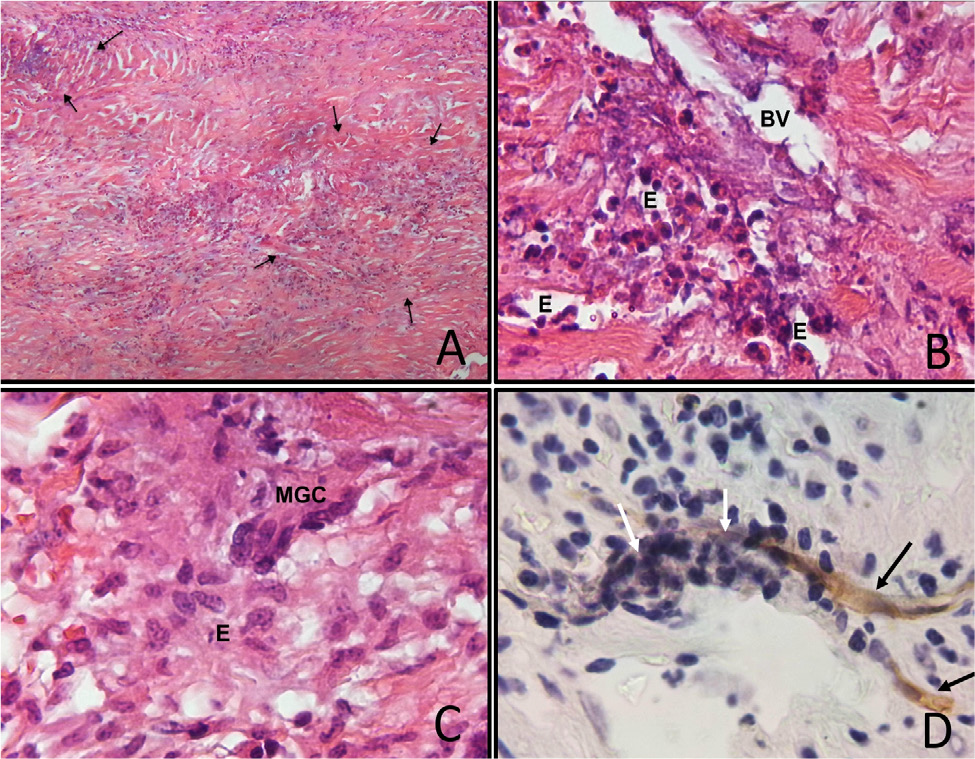
The study protocol reasonably ruled out infection, and the oedema was treated with oral steroids for three months at a decreasing dose. The headache continued, although with less intensity, and the patient was hospitalised to complete her tests. One of three cytochemical analysis of cerebrospinal fluid analysis (CSF) showed only hyperproteinorrhachia (86mg/dl); oligoclonal bands were negative. The following tests were negative or normal: CSF citology, CSF-adenosine deaminase enzyme, GeneXpert, serum antinuclear antibodies, antineutrophil cytoplasmic antibodies (ANCA) and G4-immunoglobulin, serum angiotensin converting enzyme, surface electroencephalogram, and an ultrasound on thyroid and parotid glands. Auditory evoked potentials showed severe peripheral, bilateral and symmetrical dysfunction of the auditory pathway with absolute blockage. Fludeoxyglucose-18F positron emission tomography demonstrated an intense decrease in uptake at the right temporal cortical level, and ipsilateral parietal cortical hypometabolism was found. Dura matter biopsy showed patches with increased cellularity that contrasted with areas of normal appearance. In the cellular areas, necrotising vasculitis and peripheral inflammatory infiltrate made up of accumulation of epithelioid cells, occasional multinucleated giant cells and abundant eosinophils were observed (Fig. 2). Immunohistochemical staining for CD34 showed remnants of endothelium amid necrosis and acute inflammation. A final diagnosis of EGPA was made.
(click to enlarge)
Figure 2. A) Dura mater exhibited patches with increased cellularity (arrows); B) Necrotizing vasculitis with eosinophils (E) in a blood vessel (BV); C) Accumulations of epithelioid cells (E), and occasional multinucleated giant cells (MGC); D) CD34 immunohistochemical staining showed remnants of endothelium (black arrows, brown stain) amid necrosis and inflammation (white arrows).
During her hospitalisation, the patient received a methylprednisolone bolus scheme of 1 g daily, for five days. She was discharged with 10 mg of prednisone every 24 hours, with a plan for two years of treatment under periodic monitoring. She was examined in the outpatient clinic every month for the first four months, and then every two months for a period of eight months, with adequate adherence to treatment, and without apparent adverse effects. The headache practically disappeared with the treatment, but the hearing impairment remained unchanged. During the monitoring time, there were no relapses or new symptoms. Occasionally she reported mild frontal headache, which responded to non-steroidal analgesics.
DISCUSSION
HP could be the manifestation of ANCA-associated vasculitis, that includes microscopic polyangiitis, granulomatosis with polyangiitis (GP) and EGPA; these disorders are classically systemic[1]. Our patient presented HP with EGPA, which apparently started clinically with a headache, although she had previously developed bilateral symmetrical hearing loss. In addition to HP, severe cerebral and cerebellar oedema was documented. However, ANCA were negative. Indirect immunofluorescence discriminates the perinuclear pattern (p-ANCA) and the cytoplasmic pattern (c-ANCA); the enzyme-linked immunosorbent assay technique detects antibodies against myeloperoxidase (MPO) and proteinase 3 (PR3). Most p-ANCA are anti-MPO and most c-ANCA are anti-PR3[1]. However, 50% of patients with EGPA are seronegative, 5% have antibodies against proteinase-3 and 45% against myeloperoxidase. In general, patients with negative serology have less severe systemic disease, or one limited to a single organ[1].
There is an increased risk of developing EGPA in HLA-DRB1 subjects, especially with the *04 and *07 alleles and the HLA-DRB4 gene; there seem to be two types of genetically distinct subgroups of EGPA – ANCA-positive and negative subgroups. There is a strong association of the MPO+ subset with HLA-DQ; the ANCA-negative group has a mucosal/barrier dysfunction origin associated with variants at the GPA33 gene (it encodes a cell surface glycoprotein that maintains barrier function in the intestinal epithelium) and the IL5/IRF1 gene[2]. These genetic characteristics were not investigated in our patient.
Patients with HP frequently report an oppressive, chronic, holocranial headache, sometimes associated with neurological manifestations such as cerebellar ataxia, cranial neuropathies (most frequently, optic neuropathy) and seizures[1]. The patient had hearing loss without vestibular symptoms. Sensorineural hearing loss (unilateral or bilateral) has been reported to occur occasionally with or without vestibular symptoms; the capture of nerves in the internal auditory canal and secondary neuronal damage are suspected to be the causes of hearing and vestibular loss. The other symptoms are explained by the dura mater vasculitis[1].
Meningeal thickening with contrast medium enhancement is identified in the MRI study; vasogenic oedema can be observed, frequently associated with venous thrombosis, but there may be significant oedema without it, as in our patient[3]. The diagnosis is established with a biopsy that shows necrotising vasculitis and peripheral accumulation of eosinophils and extravascular granulomas, as demonstrated in the patient[1]. There have been previous reports of EGPA with negative serology and HP, such as a 42-year-old man with asthma[4], a 47-year-old female patient with chronic sinusitis and HP, and a 78-year-old man with left temporal headache, pulmonary infiltrates, diplopia, sinusitis and hypereosinophilia[5]. But as far as we know, this is the first case of HP with EGPA histology, ANCA-negative serology and a lesion limited to the meninges and brain. This case is important because it shows that EGPA is probably a spectrum of diseases where systemic involvement predominates, but there may be cases where there is histological evidence, without the systemic context or positive serology. It also emphasises the importance of a biopsy to reach a definitive diagnosis. To the extent that more cases – adequately documented – are reported, the perspective of vasculitis that starts in the central nervous system will probably expand.
In these cases, there is no consensus on treatment; however, the first option is initially high-dose corticosteroids and later on, maintenance with oral steroids. In cases refractory to steroid treatment, the use of rituximab has been suggested.
In the few cases reported in the literature, the prognosis for life and function have been moderately good. The patient did not have hearing recovery, but no relapses or new symptoms have been documented in a period of 18 months.
CONCLUSION
Vasculitis limited to the central nervous system is rare. Vasculitis is generally reported as part of a systemic disease; however, our patient falls outside the known standards, since she presented seronegative vasculitis limited to the central nervous system, with initial manifestation of sensorineural hearing loss. This was followed by HP and hemispheric vasogenic oedema, without venous thrombosis. A biopsy showed small vessel disease with necrotising vasculitis, and granulomas and eosinophils; therefore, it is possible that we are facing a spectrum of eosinophilic GP, breaking the paradigm of only systemic possibility.