ABSTRACT
Background: Anti-leucine-rich glioma inactivated 1 limbic encephalitis (anti-LGI1 LE) is one of the most frequent autoimmune encephalitis, commonly coexisting with other autoimmune diseases. Rheumatoid arthritis (RA) and monoclonal gammopathy of unknown significance (MGUS) are commonly associated with autoimmune phenomena. However, neither RA nor MGUS have been described in the literature to date as coexisting with anti-LGI1 LE.
Case description: We present the case of anti-LGI1 LE in a male patient with rheumatoid arthritis, who was also found to have an MGUS. The patient was initially treated with corticosteroids and IV immunoglobulin. After a mild relapse, his treatment was complemented with rituximab, resulting in complete regression of the disease symptoms.
Conclusions: Our report provides evidence for the coexistence of anti-LGI1 LE with RA and/or MGUS, thus extending the differential diagnosis of patients suffering with these disease entities that present with neuropsychiatric symptoms suggestive of encephalitis. Moreover, this case raises challenges on the management of the coexistence of these diseases, given the lack of therapeutic guidelines and their potential interaction on a pathophysiological and a clinical level.
KEYWORDS
Anti-LGI1 autoimmune encephalitis, limbic encephalitis, rheumatoid arthritis, MGUS, hyponatraemia
LEARNING POINTS
- In a patient with known autoimmune or malignant background who presents with neuropsychiatric symptoms, after excluding infectious encephalitis or central nervous system involvement in the primary disease condition, autoimmune limbic encephalitis (LE) should also be considered.
- In a patient diagnosed with anti-LGI1 LE there should be an extensive check for coexisting occult pre-malignant conditions, even for months after disease presentation.
- Clinical management and treatment options of anti-LGI1 LE when coexisting with other autoimmune or pre-malignant conditions can be challenging; thus, more research is needed towards that direction.
INTRODUCTION
Anti-leucine-rich glioma inactivated 1 limbic encephalitis (Anti-LGI1 LE) is characterised by the presence of specific autoantibodies that target the LGI1 protein, a neuronal surface-exposed domain of the voltage-gated potassium channel (VGKC) complex[1]. It is one of the most frequent autoimmune encephalitis (AE), and the most frequently reported AE coexisting with autoimmune diseases although, to our knowledge, neither with rheumatoid arthritis (RA) – one of the most prevalent chronic systemic autoimmune diseases[2] – nor with haematological pre-malignant conditions. Here, we describe a case of anti-LGI1 LE in a male patient with RA and MGUS.
CASE DESCRIPTION
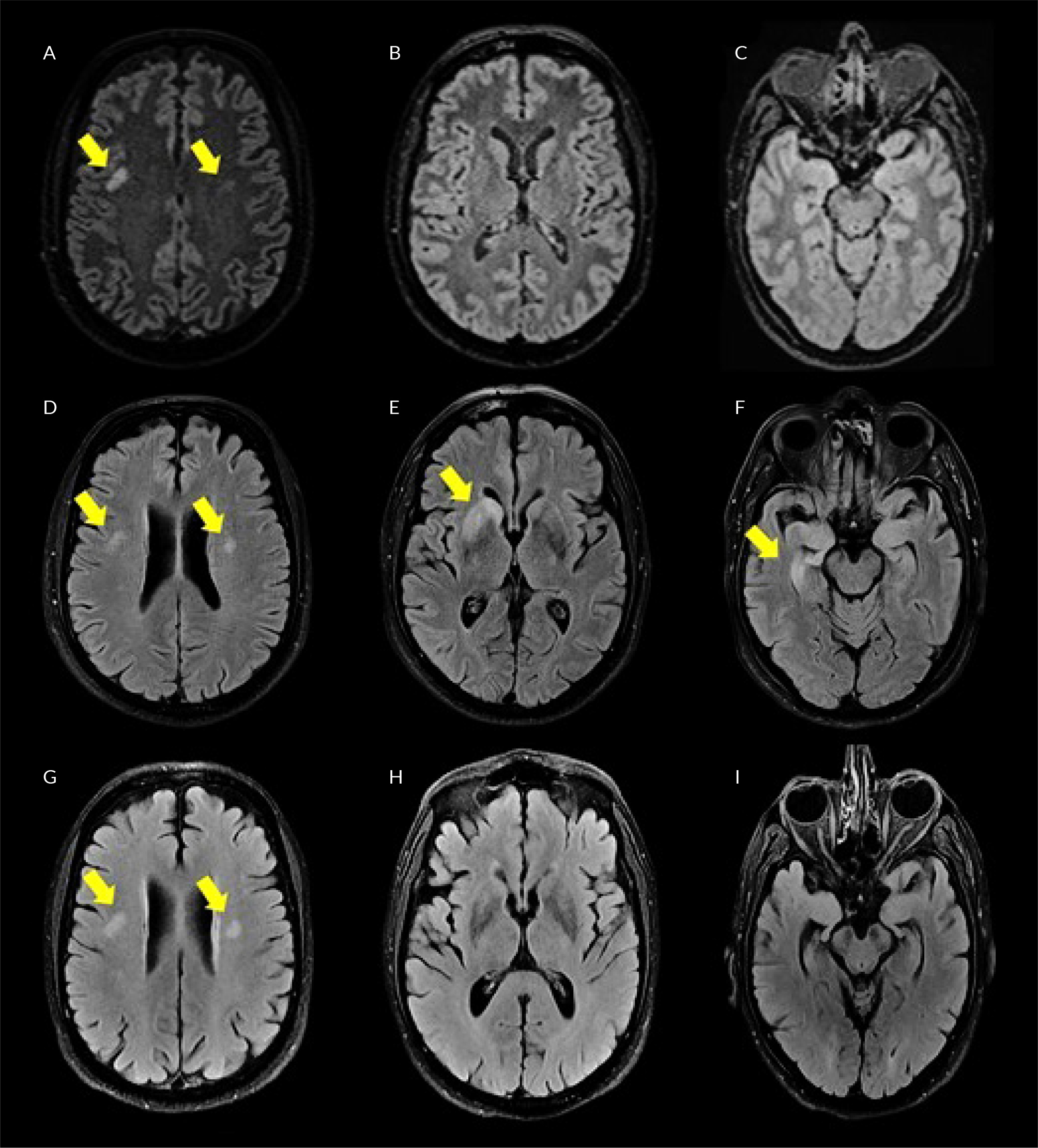
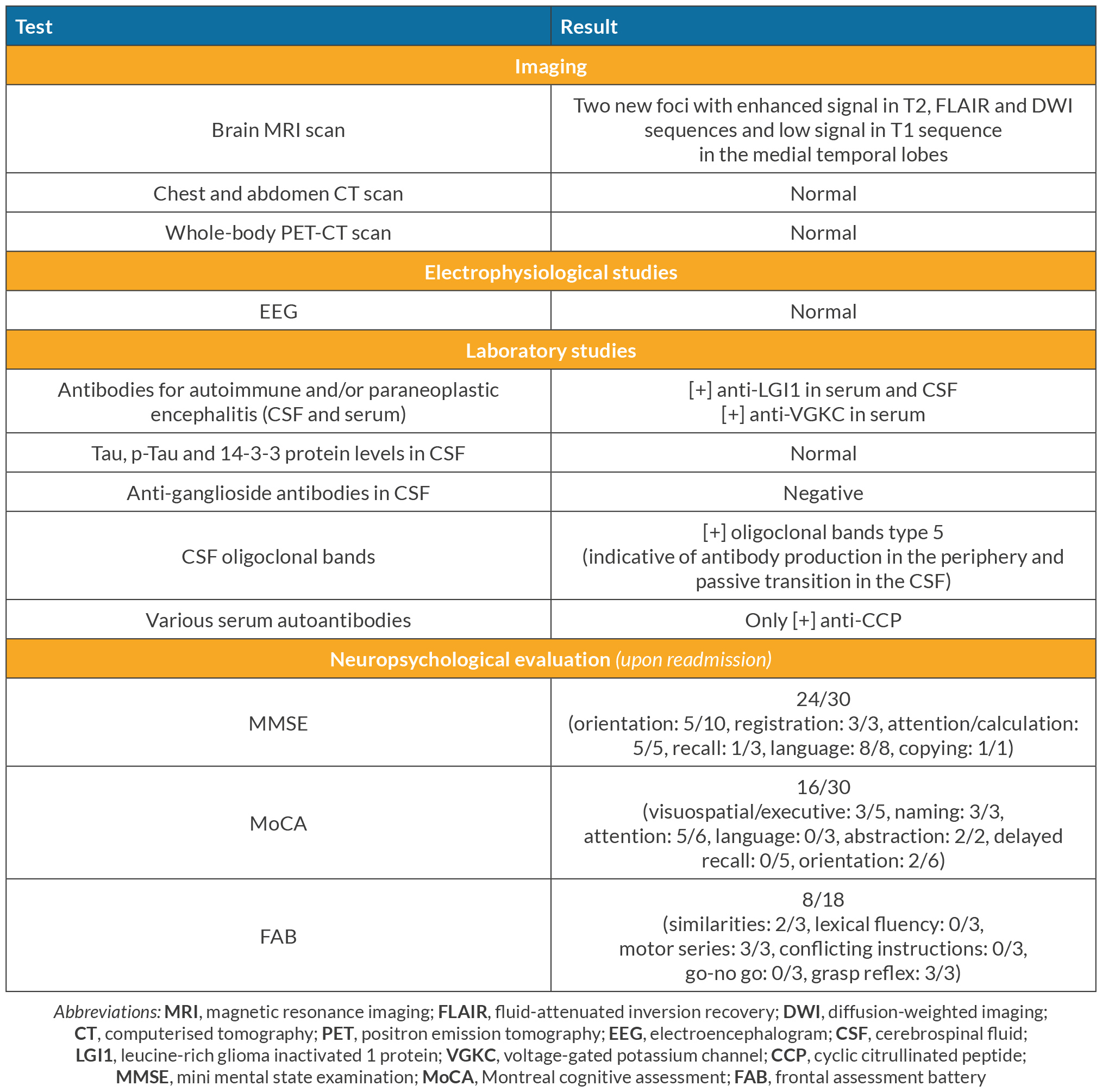
A 71-year-old Caucasian male was brought to the emergency room of our hospital after a generalised epileptic seizure that was automatically terminated, as reported by his relatives. During the last month, the patient complained of recurrent episodes of dizziness, while his relatives noticed that he seemed confused at times during the previous 10 days, with occasional episodes of short-term and episodic memory loss and disorientation. His medical history included RA (in chronic remission under leflunomide), arterial hypertension, hyperlipidaemia, thyroidectomy (due to multiple thyroid nodules 20 years ago) and benign prostate hyperplasia. On admission, the patient was alert, presenting slow cognitive activity, oriented in place-time-people, with normal vital signs (BP 115/70 mmHg, HR 70 bpm, RR 12/min, temperature 36°C, SaO2 97%, FiO2 0.21) and no focal neurological deficit. The initial brain imaging (CT and MRI scans) was normal, besides two old non-specific nodular foci on the posterior border of both frontal lobes, without contrast enhancement in MRI T2 sequence (Fig. 1A–C). A lumbar puncture (LP) was performed that showed 37 cells, all lymphocytes, with normal protein and glucose levels. Given the negative Gram stain, India ink stain and film array (which detects a series of most common bacterial and viral central nervous system pathogens), the patient was started on IV acyclovir 10 mg/kg of body weight (BW) three times per day, for the treatment of a probable viral encephalitis. Anti-seizure therapy with levetiracetam 1,000 mg twice daily was added. Meanwhile, the patient showed a progressively worsening hyponatraemia with high levels of urine sodium (Na) (serum Na upon admission 133 mmol/l, lowest serum Na 122 mmol/l, urine Na 140 mmol/l, FENa 1.1%), compatible with either syndrome of inappropriate antidiuretic hormone secretion, or salt wasting syndrome. The patient was treated with water restriction and small volumes of hypertonic saline, levetiracetam was changed to brivaracetam and hyponatraemia slowly resolved (serum Na 139 mmol/l and urine Na 63 mmol/l upon discharge). After 14 days of hospitalisation, the patient was discharged while being afebrile, oriented, with no recorded or reported new episodes of memory loss, confusion or disorientation. A third LP performed three days before discharge revealed no cells and normal glucose and protein levels. The patient was instructed to complete 21 days of antiviral treatment in total with valacyclovir per os (PO), and to continue brivaracetam as well as his previous medication. Eight days after discharge, the patient was readmitted to our hospital due to gradually worsening episodic memory, visual hallucinations, paraphasia and involuntary sudden, brief, asymmetric, repetitive (more than 100/day, even during sleep) myoclonic-like movements of his limbs and head, compatible with faciobrachial dystonic seizures (FBDS). His follow-up laboratory results revealed recurrence of hyponatraemia (Na 126 mmol/l). A new LP was performed that revealed 30 lymphocytes with, once again, normal protein and glucose levels. Consequently, the patient was restarted on IV acyclovir 10 mg/kg BW three times daily, as a recurrent viral encephalitis. The new cerebrospinal fluid (CSF) film array, serological testing for West Nile virus and Gram and Indian ink stains were all negative. Two days later, a second LP detected no cells in CSF, with normal glucose and protein. Given the patient’s progressively worsening neuropsychiatric condition, a possible diagnosis of paraneoplastic or AE, toxic encephalopathy, vascular or neurodegenerative disease was considered, so an extensive paraclinical workup was performed (Table 1). Given the CSF finding of type 5 oligoclonal bands, indicative of antibody production in the periphery, further testing with serum protein electrophoresis and immunofixation was performed, which revealed the presence of two monoclonal IgG fragments. In turn, a bone marrow biopsy was performed that exhibited 8% infiltrating plasma cells, compatible with monoclonal gammopathy of unknown significance (MGUS). The patient’s laboratory tests revealed positive anti-LG1 antibodies in serum and CSF. With the subacute onset of memory deficits, FBDS and apathetic behaviour, the T2-weighted FLAIR MRI brain abnormalities in the medial temporal lobes, the CSF pleocytosis and hyponatraemia, this led to the diagnosis of anti-LGI1 LE, in accordance with the proposed criteria[3]. The patient was started on IV methylprednisolone 1 gm/day for 5 days and IV immunoglobulin (80 gm divided over three days and 10 gm divided over three days one month later), followed by PO methylprednisolone 48 mg/day for four weeks. The patient showed a slow but stable improvement on the neurophysiological examination during the following days, with a mini mental state examination (MMSE) of 30/30, Montreal cognitive assessment (MoCA) of 20/30 and frontal assessment battery (FAB) of 8/18 at day 8, compared with 24/30, 16/30 and 8/18 respectively on day 1 of readmission (the patient’s educational baseline was 12 years of education), while serum sodium levels normalised. The patient was discharged on a PO corticosteroid tapering schedule and close follow-up.
(click to enlarge)
Figure 1. Brain FLAIR-weighted MRI imaging (horizontal plane) showing the progression and remission (after beginning of immunosuppressive treatment) of lesions due to anti-LGI1 limbic encephalitis (LE). A-C) initial presentation; D-F) first readmission and initial diagnosis of anti-LGI1 LE; G-I) 10 days after the initial treatment with IV corticosteroids and immunoglobulin; A, D and G) initial-old lesion bilaterally; B, E and H) the presence (E) and remission (H) of inflammatory lesions in the medial temporal lobes (next to the frontal horns of the lateral ventricles), predominantly in the right one. C, F and I) the presence (F) and remission (I) of inflammatory lesions in the medial temporal lobes at the level of basal ganglia between the caudate lobe and the lentiform nucleus.
(click to enlarge)
Table 1. Workup performed during readmission for the differential diagnosis of encephalopathy.
Despite having a normal electroencephalogram (EEG) and brain MRI 10 days after his latest discharge (Fig 1G–I), the patient was readmitted to our hospital 1.5 months later due to apathetic behaviour and dysthymia, as diagnosed during his outpatient follow-up. The new CSF studies showed no cells, and normal glucose and protein levels. Having high awareness of the disease’s high relapse potential[4], the patient was then started on IV rituximab (1 gm divided into two doses, 15 days apart) complementary to the PO methylprednisolone, and was discharged on regular follow-up on an outpatient basis. During his follow-up the patient significantly improved, the neurological examination was normal, and the anti-LGI1 antibody levels significantly decreased in both CSF and serum. MRI imaging showed no new contrast-enhanced focus and the neuropsychological examination showed stabilisation with minimal cognitive deficit regarding visuospatial memory, immediate and delayed recall, and synergy (MMSE: 30/30, MoCA: 20/30, FAB: 13/18).
Six months later, despite the initially negative malignancy workup (whole body CTs and PET scan), a prostate biopsy was performed, due to rising prostate-specific antigen serum levels, revealing a localised prostate cancer (Gleason score 6 (3+3), T2N0M0), for which the patient underwent a successful robotic prostatectomy. During the latest neurological evaluation, one year after the diagnosis of anti-LGI1 LE, the patient was clinically stable, with no changes on his performance on the neuropsychological tests.
DISCUSSION
In this report, we present a case of anti-LGI1 LE in a male patient with RA, a newly diagnosed prostate cancer and MGUS. The diagnosis of the anti-LGI1 LE was based on a typical clinical, laboratory and imaging presentation of the disease[3], none of which was present on initial presentation. However, the presence of hyponatraemia of non-specific cause and a referred epileptic generalised seizure can be suggestive of the presence and the subacute onset of this LE, though not pathognomonic.
Anti-LGI1 LE is one of the most common types of AE, with high coexistence rate with other autoimmune diseases. More specifically, in a retrospective cohort study of 517 patients with AE, anti-LGI1 LE was most frequently reported to coexist with systemic autoimmune diseases at a higher frequency, when compared with those with anti-NMDAR LE (the most frequent LE). Moreover, no correlation between the presence or type of autoimmune disease and the AE clinical course was noted[2]. Interestingly, RA was not among the reported autoimmune diseases found in patients with AE.
Accordingly, RA has been associated epidemiologically, and/or pathophysiologically, with several other autoimmune diseases both systemic and organ-specific, but not with anti-LGI1 LE[5]. Since the potential clinical consequences of coexistence of anti-LGI1 LE with RA remain vague, this poses a therapeutic challenge in terms of selecting the right regimen for both autoimmune diseases. To treat both RA and LE, we commenced intravenous rituximab after a clinically established relapse of neuropsychiatric symptoms, since rituximab is a recommended second-line treatment for RA[6] and has also been tested in LE cases, compared with the first-line immunosuppressive treatment options[7].
Furthermore, the malignancy workup of our patient revealed the presence of MGUS and localised prostate cancer (months after initial presentation). Regarding this coexistence of anti-LGI1 LE with malignancy, there is no relevant report involving haematological pre-malignant states such as MGUS[8], whereas only a few cases of solid tumours have been reported; for prostate cancer, in a retrospective study of 76 patients with LGI1 antibody-related cognitive deterioration, two out of five patients with malignancy had a prostate tumour[9].
CONCLUSION
Our report provides the first evidence regarding the coexistence of anti-LGI1 LE with RA and MGUS, and the accompanying diagnostic and treating challenges. Given the lack of available clinical data on the potential interference of RA or MGUS on the clinical course of LE and the lack of any prior knowledge on combined therapeutic recommendations, more research is needed towards defining the potential interactions establishing the appropriate clinical management.