ABSTRACT
Background: Vaccine-induced immune thrombotic thrombocytopenia (VITT) is a rare life-threatening thrombotic reaction to COVID-19 vaccines.
Case description: Two young male first cousins, with a family history of idiopathic thrombocytopenic purpura, developed VITT after the Ad26.COV2.S vaccine. Both had a favourable clinical and analytical outcome. We investigated the genetic factors that could be associated with a genetic predisposition to VITT.
Conclusions: There are no published cases where the VITT patients were relatives. The genetic study did not reveal any likely pathogenic variants, although the prevalent polymorphism c.497A>G (p.(His166Arg)) in the FCGR2A gene was found in a homozygous state. More studies are required to better understand VITT’s pathophysiology and any underlying genetic predispositions.
KEYWORDS
COVID-19 vaccines, genetic predisposition to disease, purpura, thrombocytopenic, idiopathic, thrombocytopenia
LEARNING POINTS
- Vaccine-induced immune thrombotic thrombocytopenia (VITT), a rare but life-threatening disease, emerged with COVID-19 vaccines.
- The genetic analyses revealed the FCGR2A gene in a homozygous state.
- These cases may raise new questions about a family predisposition to VITT.
INTRODUCTION
Coronavirus disease 2019 (COVID-19) has been associated with high morbidity and mortality[1]. With unprecedented speed, vaccines against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) have been licensed and used[2].
Beginning in February 2021, several cases of unusual thrombotic events in combination with thrombocytopenia were observed in patients after vaccination with the ChAdOx1 nCoV-19 vaccine[3]. In March 2021, the same syndrome was reported with Ad26.COV2.S, another adenoviral vector-based vaccine[4]. The term vaccine-induced thrombotic thrombocytopenia (VITT) was the more widely adopted term to designate this syndrome[5,6].
VITT strongly mimics autoimmune heparin-induced thrombocytopenia (aHIT), with an unusual, severe and life-threatening thrombotic syndrome[5,6]. It promptly received attention, leading to the publication of guidelines for diagnosing and managing this disorder[6-9].
In Portugal, the vaccination against COVID-19 started in December 2020, with four types of vaccines: ChAdOx1 nCoV-19, Ad26.COV2.S, mRNA-1273 and BNT162b2. By December 2021, approximately eight million Portuguese individuals had received a vaccine dose[10]. On 2 August 2021, we identified the first VITT case at our hospital. Several weeks later, we received a second VITT patient who was a family relative of the index case.
We aim to investigate if there is a genetic predisposition to VITT. Furthermore, we believe that this report could provide new clinical information and stimulate discussion regarding the pathophysiology of VITT.
CASE DESCRIPTION
Case 1
A previously healthy 28-year-old Caucasian man received his first dose of Ad26.COV2.S on 22 July 2021 (day 0). Eight days later, he developed lower limb petechial lesions, followed by fever (38.9ºC) one day later, which subsided with acetaminophen. On 2 August (day 11), he experienced a severe headache and sought care at the emergency department.
Apart from the petechial lesions on his left lower limb, the physical examination was unremarkable; laboratory findings are detailed in Table 1. His platelet count was 24,000 cells/mm3, and the D-dimer level was elevated (>20 µg/ml; reference value <0.5 µg/ml). Other blood tests showed normal results except for elevated C-reactive protein levels. A SARS-CoV-2 reverse-transcription–polymerase-chain-reaction test on a nasopharyngeal swab returned negative.
(click to enlarge)
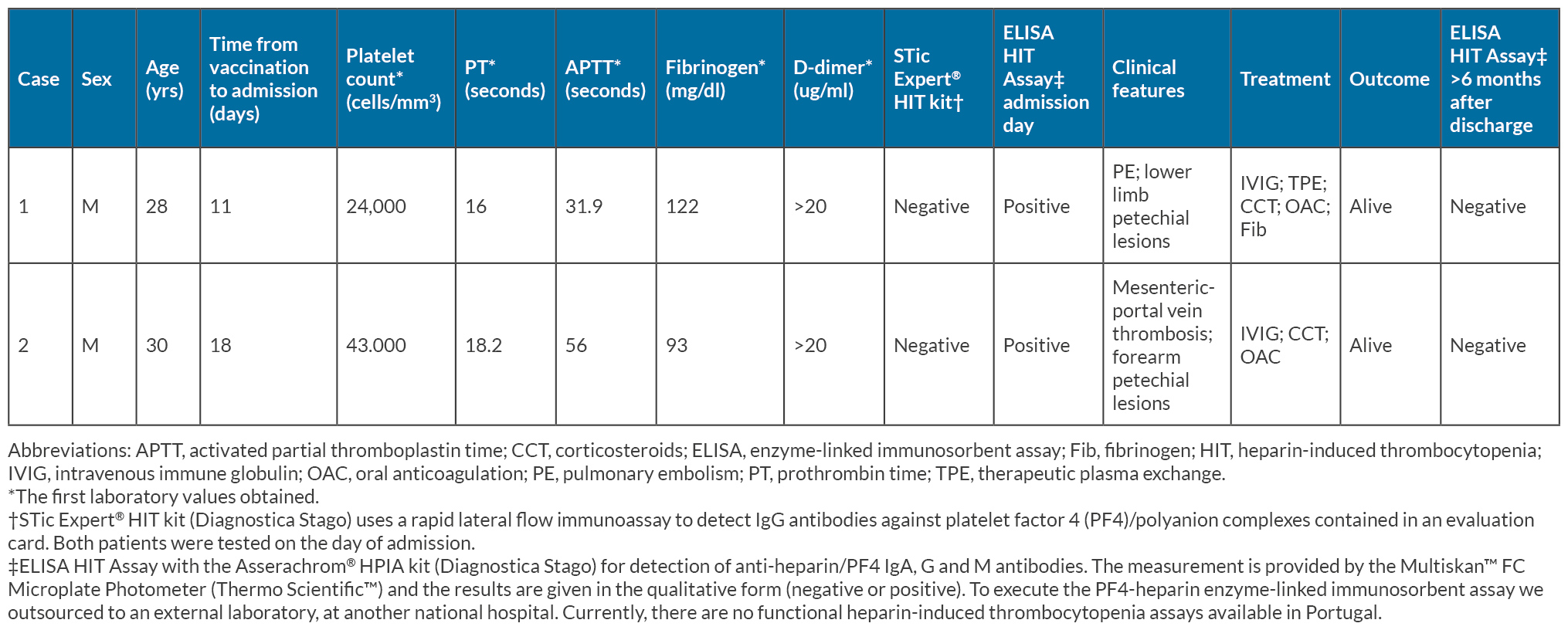
Table 1. Clinical and laboratory characteristics of the patients.
A computed tomography (CT) angiography ruled out cerebral arterial or venous thromboembolism. The patient was started on intravenous immune globulin (IVIG) at 1 g/kg/day and prednisolone at 1 mg/kg/day. Despite treatment, there was no improvement in laboratory parameters by the third day of admission, prompting a repeat CT angiography that revealed peripheral pulmonary embolism. Anticoagulation with apixaban at 5 mg twice daily was initiated. Additionally, due to a low fibrinogen value of 56 mg/dl (reference range: 200–400 mg/dL), he received 1 g of fibrinogen.
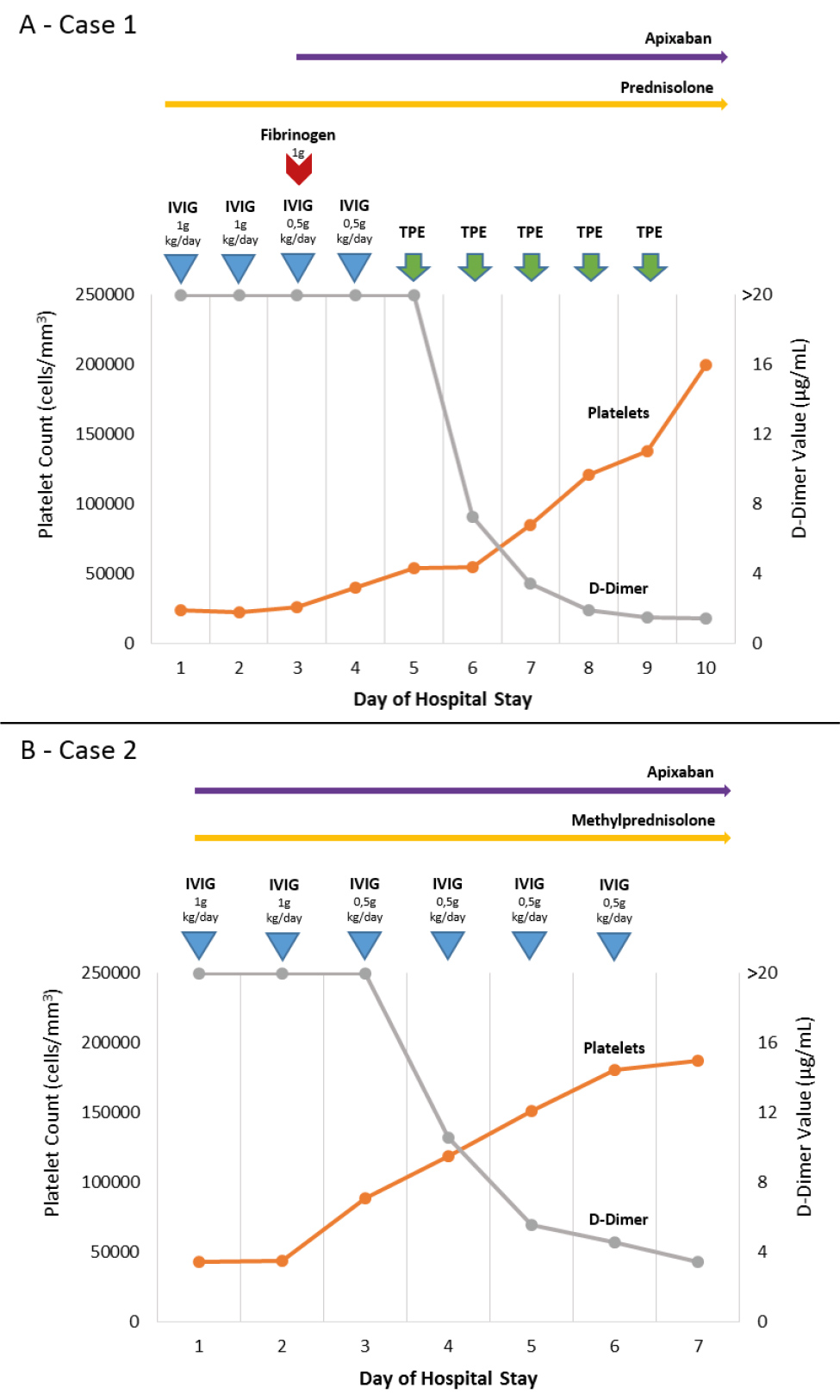
On the fifth day of admission, with persistent thrombocytopenia and high D-dimer levels despite stable clinical status, a multidisciplinary team opted for therapeutic plasma exchange using full plasma. Apixaban plasma levels were monitored before and after the procedure, showing no significant changes. The patient responded well to treatment, as depicted in Fig. 1, with progressive normalisation of platelet count and coagulation parameters. Upon discharge, he continued to be monitored and in June 2022, his platelet count remained normal with negative anti-heparin/platelet factor 4 (PF4) antibodies, nine months after corticosteroid tapering.
Case 2
A previously healthy 30-year-old Caucasian man received his first dose of Ad26.COV2.S on 1 August 2021 (day 0). Over the following two days, he reported experiencing minor symptoms such as fatigue and myalgia. Eight days later, he developed a headache and fever. While the fever responded to ibuprofen, the headache persisted. On 13 August (day 12), he began to experience upper abdominal pain that did not responded to analgesia, prompting him to visit the emergency department on 19 August (day 18).
Apart from small petechial lesions on his right forearm, the physical examination was unremarkable. Laboratory results are detailed in Table 1, showing a platelet count of 43,000 cells/mm3 and a D-dimer level exceeding 20 µg/ml (reference value <0.5 µg/ml). Other blood tests yielded normal results except for elevated C-reactive protein levels. A SARS-CoV-2 reverse-transcription–polymerase-chain-reaction test on a nasopharyngeal swab returned negative.
A CT angiography revealed mesenteric and portal vein thrombosis while ruling out arterial or venous cerebral and pulmonary thromboembolism. Treatment was initiated with IVIG at 1 g/kg/day, methylprednisolone at 1 mg/kg/day and apixaban at 5 mg twice daily. The patient responded well to treatment, as depicted in Fig. 1.
(click to enlarge)
Figure 1. Platelet count and D-dimer value evolution with the instituted therapy. Abbreviations: IVIG, intravenous immune globulin; TPE, therapeutic plasma exchange.
He experienced a favourable clinical and analytical outcome with progressive normalisation of platelet count and coagulation parameters. Following discharge, he continued to be monitored, with the last evaluation in July 2022 showing a normal platelet count and negative anti-heparin/platelet factor 4 antibodies, eleven months after corticosteroid tapering.
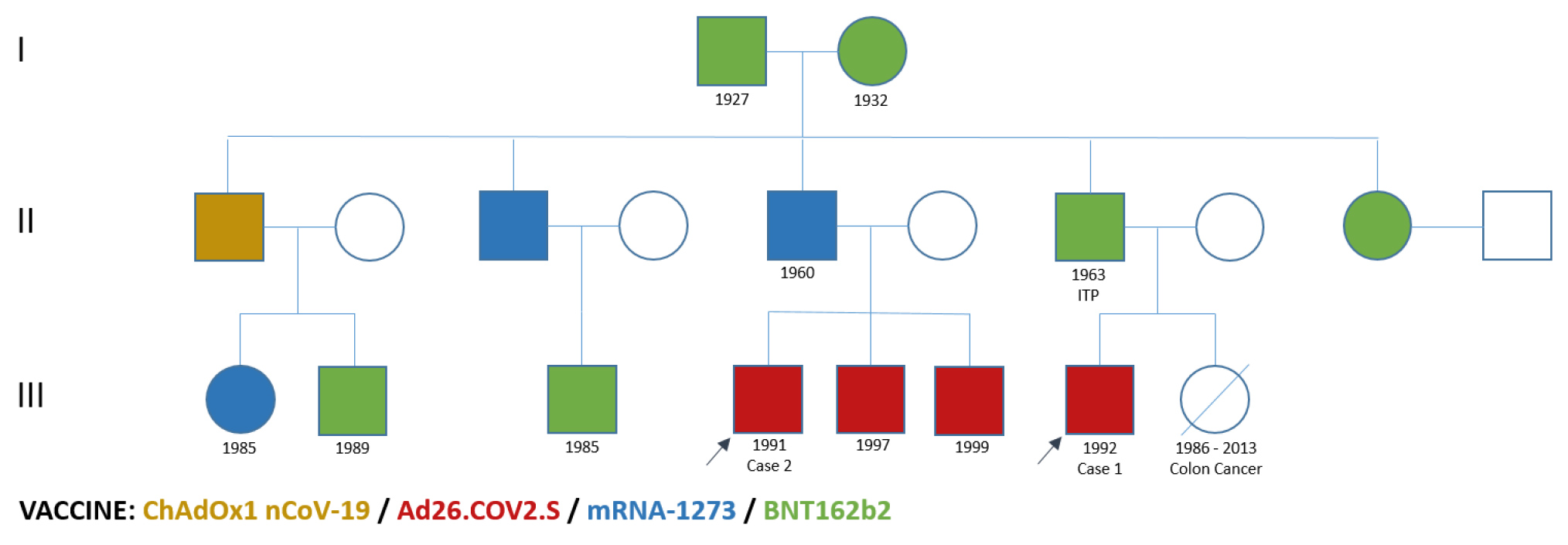
This patient is the father’s cousin of the index case. His uncle, who is the father of Case 1, has a history of idiopathic thrombocytopenic purpura (ITP) since childhood, necessitating splenectomy. The family history of these patients was evaluated (Fig. 2), revealing no other family members with a history of haematologic diseases. Three other family members had received adenoviral vector-based vaccines before the onset of the family VITT cases. No other relatives reported any vaccine complications beyond minor symptoms.
(click to enlarge)
Figure 2. Family vaccine history. Abbreviations: ITP, idiopathic thrombocytopenic purpura.
Despite international recommendations[8,9], our patients opted not to receive a booster mRNA vaccine dose. Following the occurrence of the family VITT cases, the other family members received a non-adenoviral vector-based booster vaccine dose, without experiencing any vaccine complication beyond minor symptoms.
DISCUSSION
With the successful COVID-19 vaccination programme, VITT has emerged as a rare yet severe complication. The clinical and laboratory features of VITT, which closely resemble autoimmune HIT, are believed to stem from the production of anti-PF4 antibodies in response to vaccine components. However, the specific components involved and the exact pathophysiology remain unclear[1,6].
In general, the clinical, laboratory and pathological characteristics of our patients were consistent with the literature[12]. Both individuals experienced a positive clinical and analytical outcome, although the index case required therapeutic plasma exchange. Notably, studies indicated a slightly higher occurrence of VITT in women, whereas our patients were male, related by family and younger than the median age at presentation (48 years)[12].
There are no published cases where VITT patients were relatives (search carried out on Medline/PubMed, Embase and Scopus with the MeSH terms COVID-19 vaccines; genetic predisposition to disease; purpura, thrombocytopenic, idiopathic; thrombocytopenia, and the keywords vaccine-induced thrombotic thrombocytopenia; family and relatives). Hence, we sought to investigate if there might be a genetic predisposition to this condition.
The clinical and laboratory features of VITT could also resemble thrombotic thrombocytopenic purpura (TTP), and acquired immunoglobulin G (IgG) mediated TTP in family members has been reported[13]. However, ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin type 1 motif, 13) activity was normal on our patients.
While genes associated with immune haematologic diseases such as ITP and HIT, as well as PF4/heparin IgG antibody levels have not been consistently identified[14,15], the FCGR2A H131R polymorphism has shown potential relevance in HIT-associated thrombosis[14,16]. The FCGR2A gene encodes the Fc gamma receptor IIa (FcγRIIa or CD32), a cell surface receptor involved in immune responses[14,16] and is believed to play a role in procoagulant pathways[6]. The immune complexes containing PF4 can signal through FcγRIIa, which generates procoagulant platelets, induces platelet/neutrophil aggregates and stimulates NETosis by neutrophils[6]. The FCGR2A H131R polymorphism is considered functionally significant in the IgG binding region[14,16] and has been associated with conditions such as ITP[15,17,18]. Genetic analyses have been performed on patients with VITT in an attempt to identify a possible role of genetic predisposition in the pathophysiology of VITT. However, the results have not been conclusive[19].
Following discussions with our geneticist team and patients, we opted to conduct a next generation sequencing whole-exome-based virtual multigene panel designed for situations of genetically determined high risk of thrombotic diseases, on Subject II-7 (Fig. 2). This patient had presented an immune haematologic disease (ITP) as described above and was the link between the two patients.
The gene panel included 41 genes (ABCG5, ABCG8, ADAMTS13, ANKRD26, CYCS, DIAPH1, ETV6, FCGR2A, FLI1, FLNA, FYB1, GALE, GATA1, GFI1B, GNE, GP1BA, GP1BB, GP9, GPR65, HOXA11, ITGA2B, ITGB3, MASTL, MECOM, MPL, MYH9, NBEAL2, ORAI1, PRKACG, PTPRJ, RBM8A, RUNX1, SLFN14, SRC, STIM1, THPO, TPM4, TRPM7, TUBB1, VWF and WAS). No (likely) pathogenic variants were found, although the highly prevalent polymorphism c.497A>G (p.(His166Arg)) in FCGR2A (NM_021642.5), classically known as FCGR2A rs1801274 H131R, was found in a homozygous state. Given its high prevalence (23% of the population is homozygous according to the gnomAD database)[20] and cost-effectiveness considerations, we chose not to pursue further genetic studies on our patients.
These cases prompt considerations about potential genetic factors influencing VITT susceptibility. Further research and time are necessary to deepen our understanding of VITT’s pathophysiology and any underlying genetic predispositions.