ABSTRACT
Background: Alagille syndrome (ALGS) is a multisystem disorder involving at least three systems among the liver, heart, skeleton, face, and eyes. Common cardiac associations include pulmonary artery stenosis/atresia, atrial septal defect (ASD), ventricular septal defect (VSD) and tetralogy of fallot (ToF). Coarctation of aorta (CoA), renal and intracranial arteries are commonly involved vessels in Alagille syndrome. We present two cases with rare cardiovascular manifestations of Alagille syndrome.
Case description: Case 1: A 25-year-old female with a history of Alagille syndrome presented to the cardiologist office for progressive exertional dyspnoea, orthopnoea, and palpitations. She was tachycardiac on examination and had an apical diastolic rumble. A transthoracic echocardiogram (TTE) showed a left ventricular ejection fraction (LVEF) of 60% and parachute mitral valve (PMV) with severe mitral stenosis. A transoesophageal echocardiogram (TOE) showed insertion of chordae into the anterolateral papillary muscle, severe mitral stenosis with a valve area of 0.7 cm. She was referred to a congenital heart disease specialist and underwent robotic mitral valve replacement with improvement in her symptoms.
Case 2: A 27-year-old female with known Alagille syndrome and resistant hypertension presented to the cardiologist office due to progressive exertional dyspnoea for a year. She was hypertensive and had a new 2/6 systolic ejection murmur along the left upper sternal border. TTE revealed an LVEF of 60% and pulmonary artery pressure of 19 mmHg. A CoA was suspected distal to the left subclavian artery due to a peak gradient of 38 mmHg. Cardiac magnetic resonance (CMR) imaging ruled out CoA, and diffuse narrowing of the descending thoracic aorta measuring 13–14 mm in diameter was noted. The patient was referred to a congenital heart disease specialist for further management.
Conclusion: PMV presenting as mitral stenosis and mid-aortic syndrome are not commonly described anomalies in association with Alagille syndrome. TTE, TOE and CMR played a key role in diagnosis and management of these patients.
KEYWORDS
Alagille syndrome, parachute mitral valve, mid-aortic syndrome
LEARNING POINTS
- Alagille syndrome (ALGS) is a complex multisystem disorder involving the liver, heart, skeleton, face, and eyes. Cardiovascular involvement occurs in up to 95% of the patients.
- Common cardiac associations include pulmonary artery stenosis/atresia, atrial septal defect (ASD), ventricular septal defect (VSD) and tetralogy of fallot (ToF).
- A parachute mitral valve (PMV) presenting as mitral stenosis and mid-aortic syndrome is not commonly described anomalies in association with ALGS. Here, we present such rare cases.
INTRODUCTION
Alagille syndrome (ALGS) is a complex multisystem disorder involving liver, heart, skeleton, face and eyes, and can be inherited with autosomal dominance with variable expressivity or can occur sporadically[1]. Mutations in JAG1 and NOTCH2 genes have been associated with this syndrome[1,2]. However, phenotypic discordance in monozygotic twins has raised suspicion of environmental factors playing a major role as well[3]. Cardiovascular involvement occurs in up to 95% of the patients and can lead to significant morbidity and mortality in Alagille syndrome patients[4]. Common cardiovascular anomalies include pulmonary atresia/stenosis, ToF and aortic stenosis. Here we describe two cases with rare findings of a parachute mitral valve (PMV) and a diffuse narrowing of the descending thoracic aorta in association with Alagille syndrome.
CASE DESCRIPTION
Case 1
A twenty-five-year-old Caucasian female with a known history of Alagille syndrome (diagnosed on liver biopsy) presented to the cardiologist office for progressive exertional dyspnoea of few months’ duration. She also had hypertension, and diabetes mellitus type 2 from focal segmental glomerulosclerosis (noted on renal biopsy), characteristic facies and a cleft palate. Dyspnoea had progressed to the point of her not being able to climb the stairs and was associated with two-pillow orthopnoea. She denied lower extremity oedema, chest pain, palpitations, or dizziness. Other than the history of liver and renal biopsies, the rest of her past medical history was unremarkable. She denied history of smoking, alcoholism, or any drug abuse. She was active in the Special Olympics and lived in a ranch with her family.
On examination in the office, the patient was tachycardiac with heart rate of 113 beats per minute, blood pressure of 125/70 mmHg, afebrile, with a respiratory rate of 14 breaths per minute, saturating 96% on room air. The physical examination was remarkable for diastolic rumble at the cardiac apex. Lungs were clear to auscultation, and the rest of the physical examination was unremarkable.
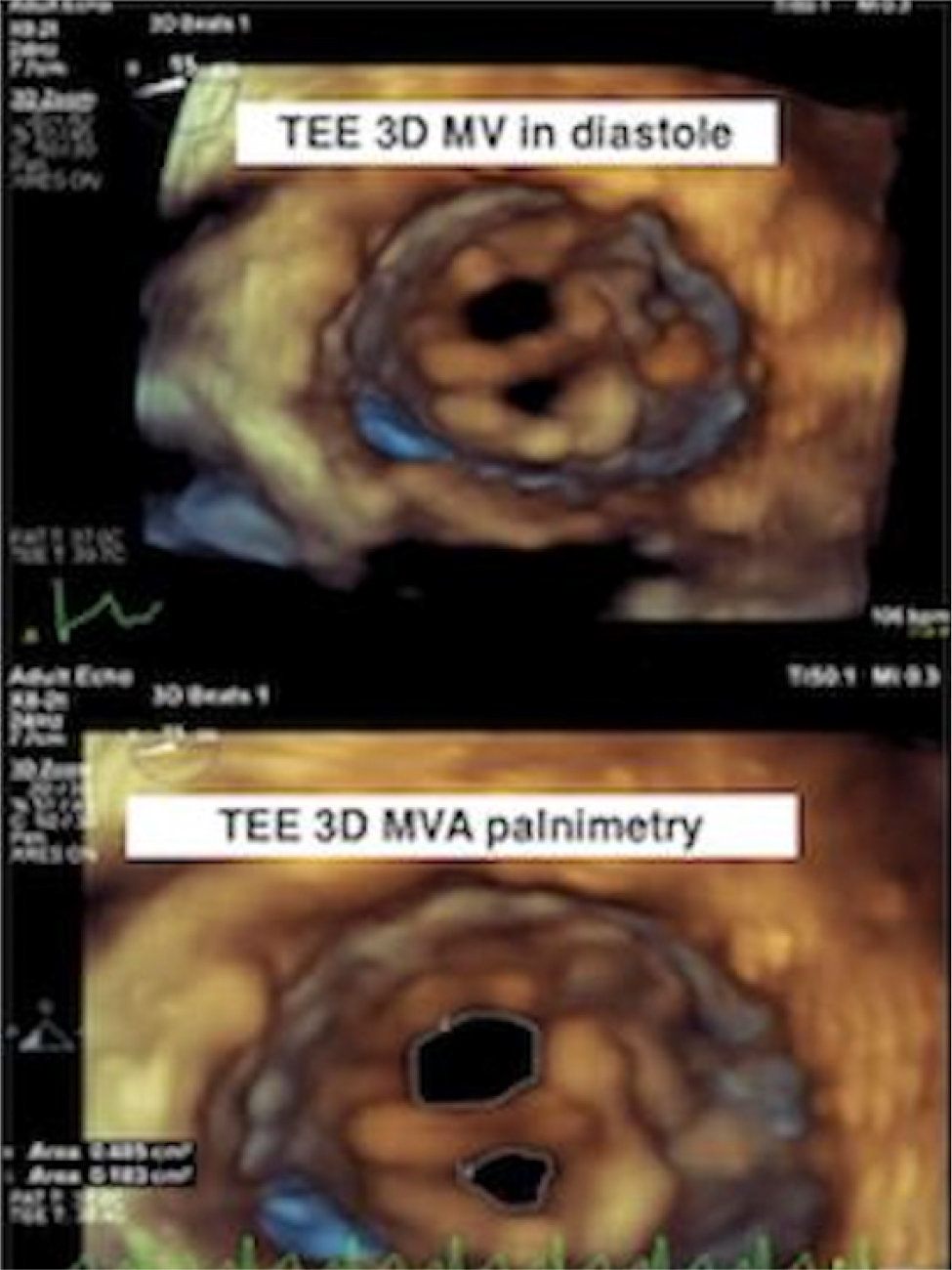
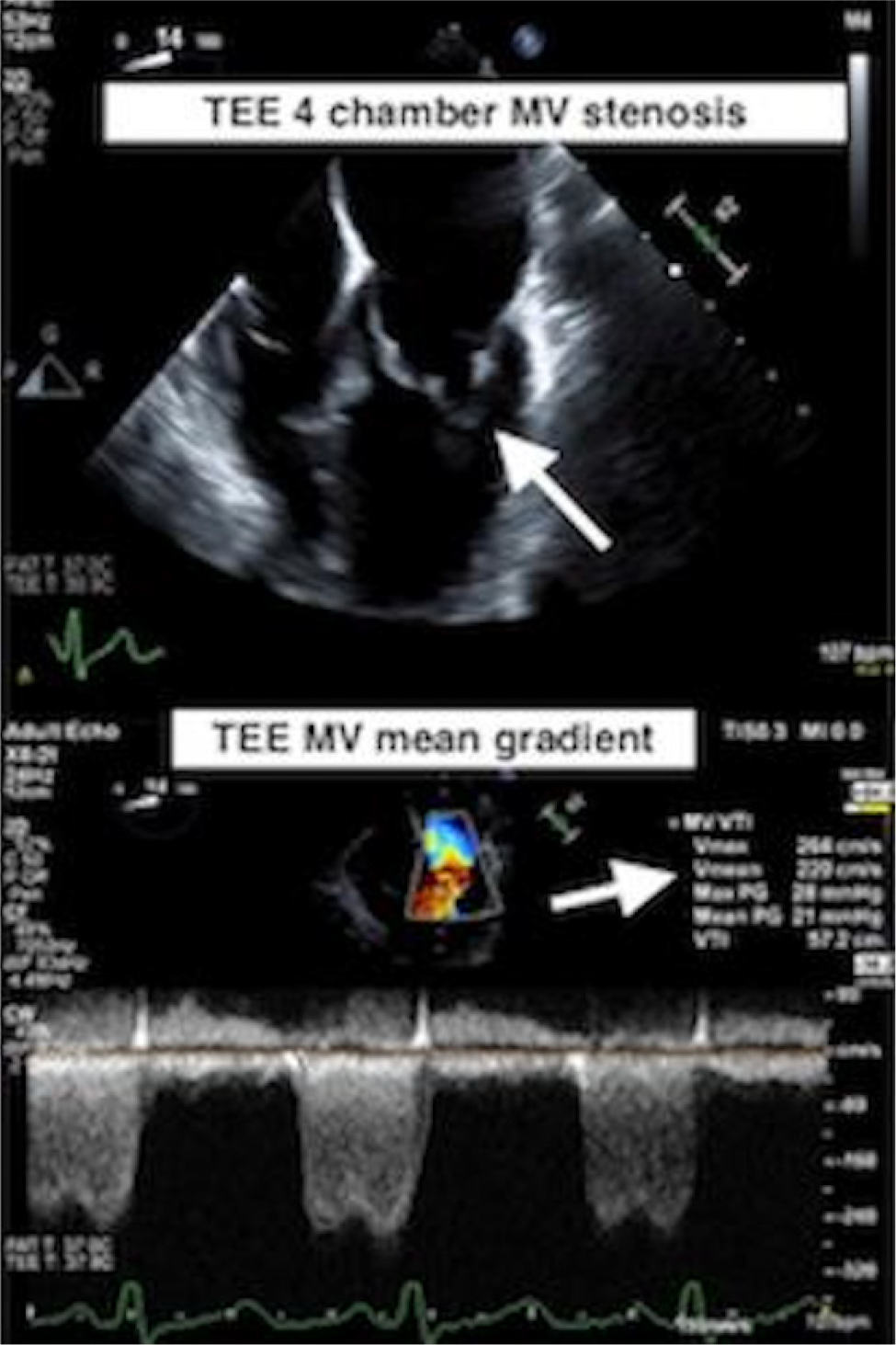
A transthoracic echocardiogram (TTE) showed a left ventricular ejection fraction (LVEF) of 60%, and a PMV with severe mitral stenosis was suspected. A transoesophageal echocardiogram (TOE) was obtained, and it revealed true PMV with insertion of chordae into the anterolateral papillary muscle (Fig. 1). There was severe mitral valve stenosis with a valve area of 4.2 cm2 by the pressure half time method. Using the continuity equation, the mitral valve area came out in the severe range of 0.7 cm2. By planimetry, the mitral valve area was 0.79 cm2 (Fig. 2). There was moderate mitral regurgitation, a physiologic amount of tricuspid regurgitation, the right ventricular systolic pressure was moderately to severely elevated and there was normal aortic arch without hypoplasia or coarctation. The patient was referred to a congenital heart disease specialist and underwent robotic mitral valve replacement with subsequent improvement in her symptoms.
(click to enlarge)
Figure 1. True PMV with insertion of chordae into the anterolateral papillary muscle on TOE.
(click to enlarge)
Figure 2. Severe mitral valve stenosis with mitral valve area of 0.7 cm2 seen on TOE.
Case 2
A 27-year-old female had history of Alagille syndrome requiring a liver transplant twice in 1996 after rejection of the first from vascular complications, a history of secondary hypertension and retinitis pigmentosa, with no known cardiac abnormalities. She presented to the office of her primary care physician for a routine annual physical examination and complained of progressive exertional dyspnoea for a year or so. She was able to walk between buildings; however, more recently, even minimal activity was causing shortness of breath and she also reported feeling “extra heartbeats” as well as occasional sharp fleeting, self-limited, non-radiating central chest pain at rest. The patient denied any peripheral oedema, orthopnoea, paroxysmal nocturnal dyspnoea or dyspnoea at rest, dizziness or loss of consciousness. Her past medical history was significant for chronic kidney disease stage 3A with baseline creatinine of 1.75 and an estimated glomerular filtration rate of 48; otherwise, it was unremarkable. There was no history of known congenital heart diseases, and no family history of premature coronary artery disease or sudden cardiac death. She lived with her parents and reported alcohol use only socially and denied use of tobacco or other recreational drugs.
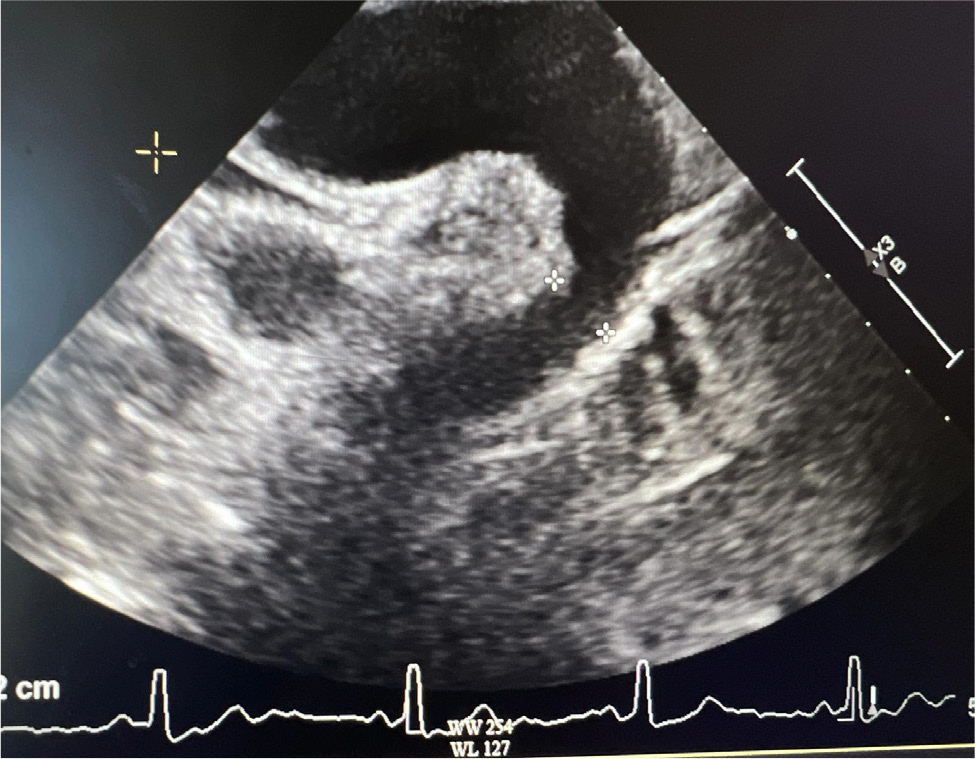
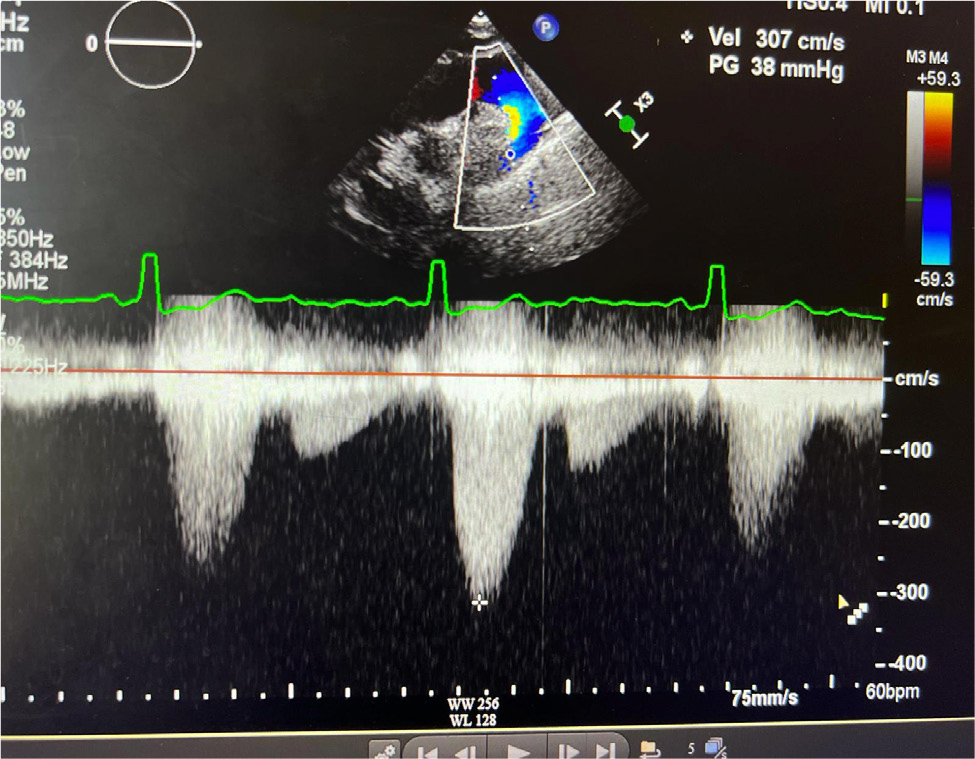
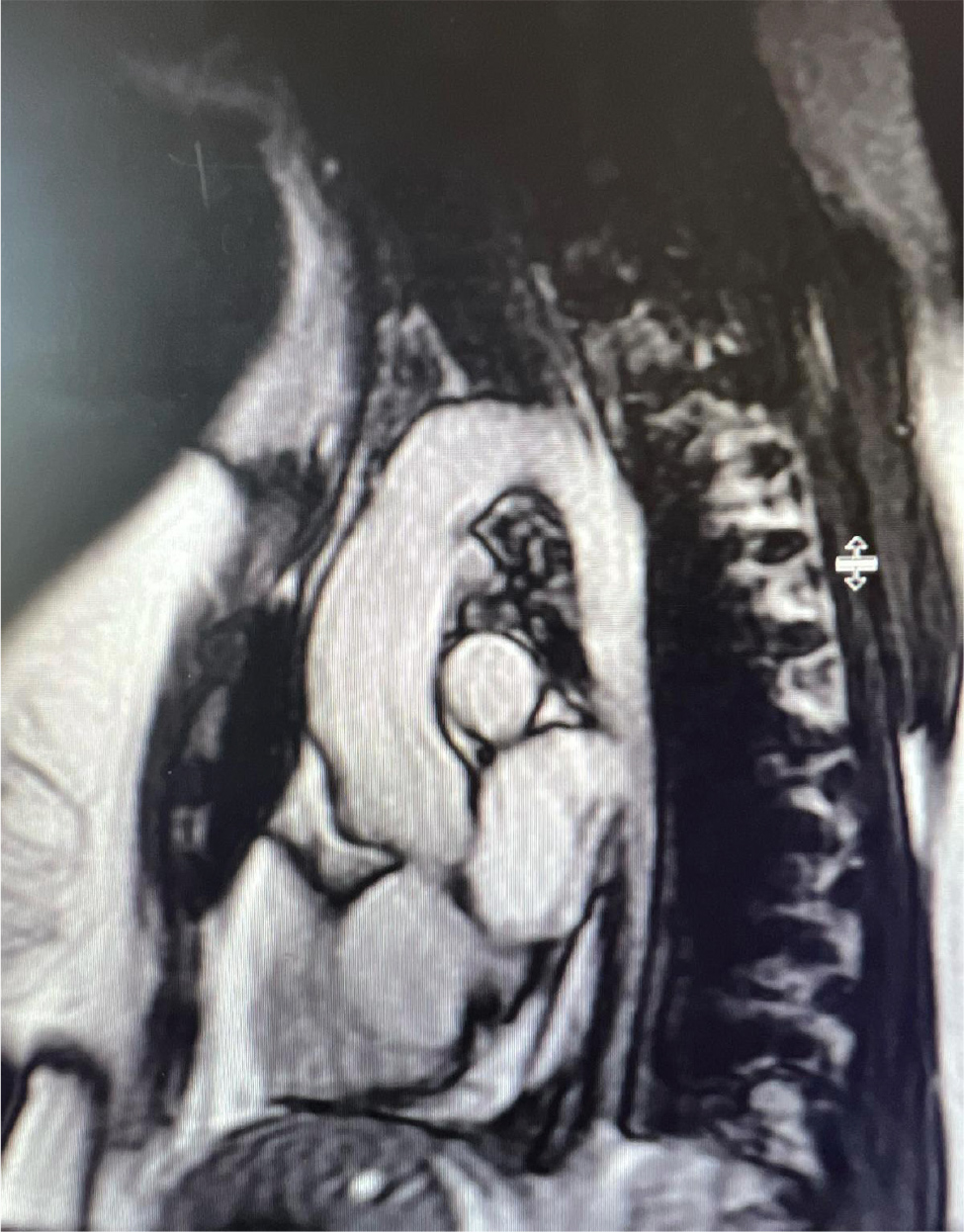
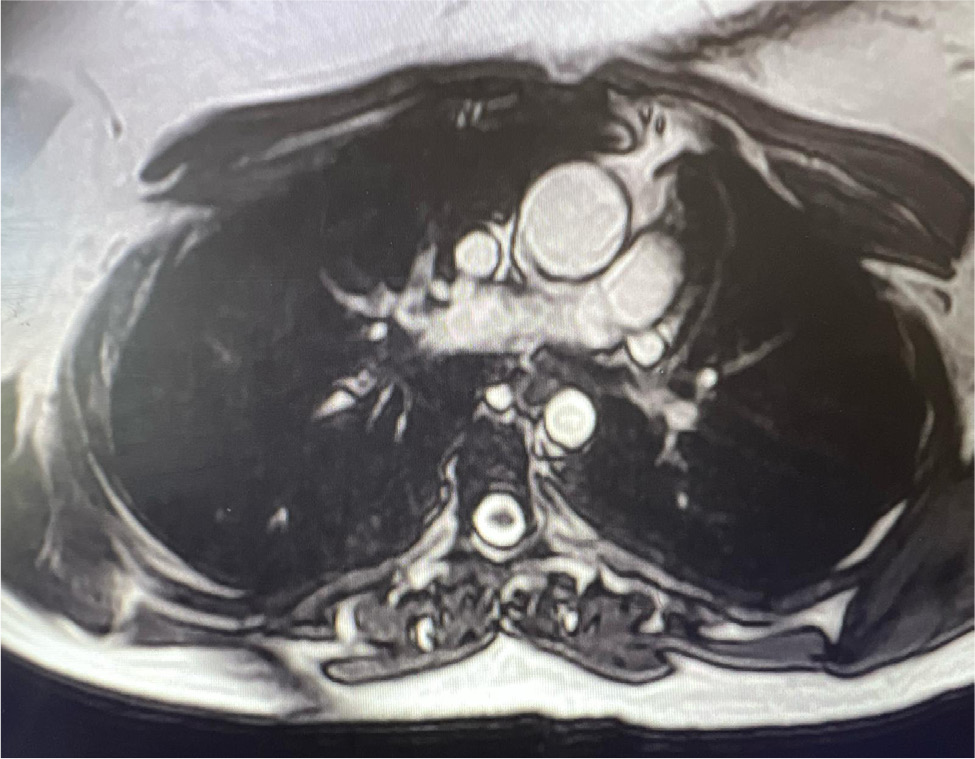
Upon arrival in the primary care physician’s office, her blood pressure was 140/70 mmHg, heart rate was 67 beats per minute, she was afebrile and saturating up to 98% on room air with respiratory rate of 16 per minute. On examination, she was not in any acute distress. She was found to have a new 2/6 systolic ejection murmur along the left upper sternal border. Her lungs were clear to auscultation, and she did not have jugular venous distension or calf swelling. The rest of the examination was unremarkable. Her laboratory test results were unremarkable, and renal function was stable at baseline. Due to clinical signs and symptoms (shortness of breath, new murmur, hypertension), her primary care physician ordered a TTE which showed an LVEF of 60–64%, with normal diastolic function. However, coarctation of the aorta (CoA) was noted distal to the left subclavian artery with a maximum gradient of 38 mmHg, and a bicuspid aortic valve was not ruled out (Fig. 3 and 4). Right ventricular systolic pressure was 19 mmHg with normal pulmonary artery pressure. Cardiac magnetic resonance imaging was ordered for further evaluation, which confirmed normal LVEF of 68% on a three-dimensional assessment. However, the patient also had severe left pulmonary artery stenosis; the diameter at the origin measured 0.2–0.3 mm with distal dilation (measuring 0.8–1 cm distally); a persistent left superior vena cava draining into coronary sinus was leading to its dilatation and there was no bridging vein. Aortic root, ascending aorta and supra-aortic vessels were normal and there was no evidence of CoA. However, mid to distal aortic arch narrowing was noted (diameter measuring 20 mm at mid-arch, and 14 mm at distal arch). There was diffuse narrowing of the entire descending aorta with the diameter measuring 13–14 mm all the way down to the level of diaphragm. The left renal artery was not well visualised. There was no evidence of ASD, VSD or patent ductus arteriosus and all four pulmonary veins were normal (Fig. 5 and 6). In summary, the findings of the left atretic left pulmonary artery were consistent with Alagille syndrome. No discrete coarctation was noted; however, there was diffuse narrowing of the thoracic aorta. Mid-aortic syndrome was suspected, and patient was referred to a congenital heart disease specialist for further management.
(click to enlarge)
Figure 3. CoA suspected on TTE.
(click to enlarge)
Figure 4. Bicuspid aortic valve on TTE.
(click to enlarge)
Figure 5. Absence of CoA confirmed on CMR.
(click to enlarge)
Figure 6. Mid to distal aortic arch narrowing seen on CMR.
DISCUSSION
Alagille syndrome is characterised by complex multi-organ involvement. Common systemic manifestations include the following[1,2]:
- paucity of intrahepatic bile ducts leading to systemic cholestasis in infancy as the most common presenting feature (up to 95% of patients);
- congenital heart disease, the second most common feature associated with this syndrome (85–95%) – peripheral pulmonary artery stenosis (most common), pulmonary artery atresia, atrial/ventricular septal defect and ToF;
- renal disease (40–73%);
- pancreatic insufficiency (approximately 40%);
- peculiar facies (77–98%), broad forehead, deep set eyes, up-slanting palpebral fissures, prominent ears, pointed chin, straight nose with bulbous tip;
- ocular embryotoxicity (61–88%) – especially posterior embryotoxin which leads to prominence of Schwalbe’s ring at the junction of iris and cornea and retinal pigment abnormalities as seen in our patient.
- growth (50–87%), and mental (0–16%) retardation and developmental delay (16–52%);
- vertebral arch defects (39–87%: butterfly vertebrae).
The five principal features of Alagille syndrome are facial dysmorphia, posterior embryotoxon, butterfly vertebrae, peripheral pulmonary artery stenosis and chronic cholestasis. Alagille syndrome diagnosis is based on the association of a paucity of bile ducts on liver biopsy with three of the five major features listed above, or with one of these features and a positive family history[5]. Both the patients described above had paucity of intrahepatic ducts diagnosed on liver biopsies. In addition, patient 1 had characteristic facies, chronic cholestasis and vertebral abnormalities. Patient 2 had retinitis pigmentosa, pulmonary stenosis and chronic cholestasis in addition to the consistent biopsy findings.
Severe cardiac disease is a significant cause of morbidity and mortality in patients with Alagille syndrome. Common cardiac anomalies associated with Alagille syndrome are isolated branch pulmonary artery stenosis or hypoplasia, ToF and left-sided lesions including valvular and supravalvular aortic stenosis[6]. ASD, VSD and hypertrophic cardiomyopathy have also been reported in association with Alagille syndrome[6,7]. Patients with combined ToF and pulmonary atresia have the highest mortality rate of up to 75% versus only 34% in patients with ToF alone[6]. Association of a PMV is a rare finding in patients with Alagille syndrome.
Vascular anomalies are common in Alagille syndrome and cause significant morbidity and mortality[1,4]. The pulmonary artery involvement is an Alagille syndrome common site; however, intracranial arteries, aorta and renal arteries, celiac, superior mesenteric and subclavian arteries are also commonly involved[4]. Common aortic involvement presents as CoA, aortic aneurysms and – rarely – as mid-aortic syndrome[4,6,8]. Mid-aortic syndrome refers to severe narrowing of abdominal aorta and ostial stenosis of its major branches including renal arteries. It has been described in association with Alagille syndrome and can cause renovascular hypertension[5,8]. Patient 2 likely had mid-aortic syndrome in the setting of Alagille syndrome, which probably caused his hypertension. However, the finding of diffuse narrowing of the descending thoracic aortic is quite a rare occurrence in such patients.
CONCLUSION
Cardiac and vascular involvement is a significant cause of morbidity and mortality in patients with Alagille syndrome. TTE, TOE and CMR can play a useful role in diagnosing these patients. Physicians should be vigilant in recognising the subtle clinical signs and symptoms so patients can be diagnosed and treated in a timely manner.