ABSTRACT
Late onset combined immunodeficiency (LOCID) is a rare variant of common variable immunodeficiency (CVID), typically affecting adult patients who present with opportunistic infections (OI) and/or low CD4+ T lymphocytes. Diagnostic delay is common due to the rareness of this entity, increasing morbidity and mortality. We report on a 66-year-old male who developed a severe gastrointestinal cytomegalovirus (CMV) infection, refractory to antiviral treatment and anti-cytomegalovirus specific human immunoglobulin administration, with a fatal outcome due to an undiagnosed LOCID.
KEYWORDS
Primary immunodeficiency, late onset combined immunodeficiency, opportunistic infection, cytomegalovirus
LEARNING POINTS
- Infections in patients with primary immunodeficiencies (PIDs) could be more severe and life-threatening than in immunocompetent hosts.
- PIDs are not exclusive to paediatric patients; diagnostic delay is common, and they should also be suspected in adulthood.
- Diagnostic delay in PID patients is associated with more morbidity and mortality.
INTRODUCTION
Infections are the most frequent clinical manifestation of patients with primary immunodeficiency (PID) and, in most cases, the first clinical manifestation[1]. Common variable immunodeficiency (CVID) is the most prevalent symptomatic PID. First described in 2009, late onset combined immunodeficiency (LOCID) is a rare variant of CVID[2]. While CVID is characterised by a defect in antibody production leading to hypogammaglobulinemia, LOCID is characterised by defects in T cells and opportunistic infections (OI)[2,3]. Lately, the DEFI study group defined LOCID patients as the ones that present OI or a naïve CD4+ T-cell count <20/μl (DEFI 2015-LOCID)[4]. Because of the severe CD4+ T-cell depletion, the impaired cellular response predisposes to viral infections such as cytomegalovirus (CMV).
CMV is a widespread virus normally causing asymptomatic infections in immunocompetent patients. Notwithstanding this, in immunocompromised hosts CMV can develop life-threatening disease due to greater severity and a poor therapeutic response[5]. The involvement of the gastrointestinal tract is frequent in CMV infection in patients with T-cell deficient response. The colon is the most frequent affected part, followed by the stomach and the oesophagus, with a diffuse affection in many cases. In cases of severe CMV infections, it can result in disseminated disease with multiorgan failure[6]. In this report, we present a PID undiagnosed host that developed a fatal CMV infection.
CASE DESCRIPTION
We report on a 66-year-old man with a history of multiple lower respiratory tract infections (LRTI) in his childhood, requiring a number of hospital admissions and developing bronchiectasis as a sequela. The patient was diagnosed with porokeratosis of Mibelli (PM) in his fifties and retinoid treatment was started by a dermatologist prescription. When he was 60 years old, due to a positive screening in the faecal blood test an endoscopic study was undertaken, demonstrating severe oesophagitis with ulcers; CMV molecular or serological tests were not performed at that time. Six years later, the patient was admitted to our hospital due to an ischaemic stroke with a National Institute of Health Stroke Scale of 14 on account of left hemiplegia with anaesthesia, limitation of left gaze and dysarthria. An occlusion of the A2 segment of the anterior cerebral artery was identified and an interventional neuroradiological procedure was indicated, achieving complete reperfusion. The family reported that the patient had suffered from constitutional syndrome in the last month, losing 5 kg in weight. The computed tomography (CT) angiography performed on admission for the neurovascular study showed nodules and ground glass over fibrosis and bronchiectasis. There was no consanguinity or immunodeficiency history in the family.
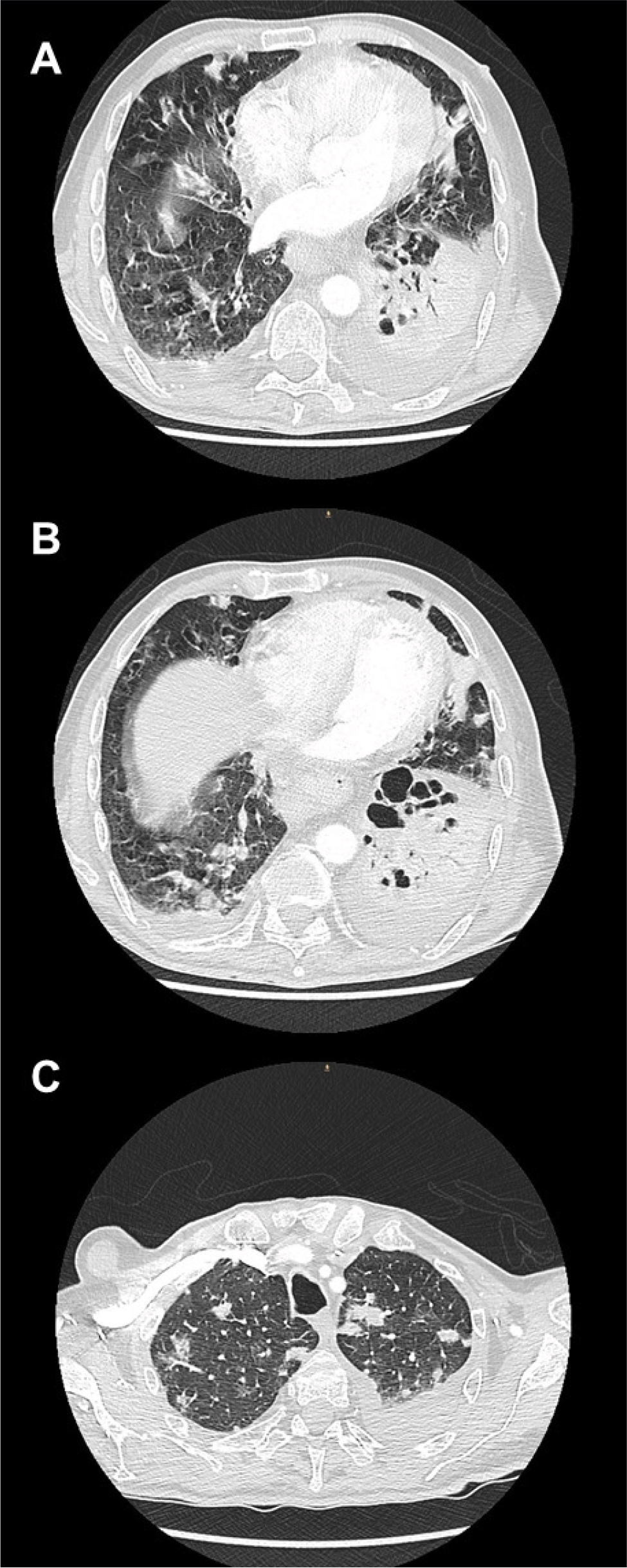
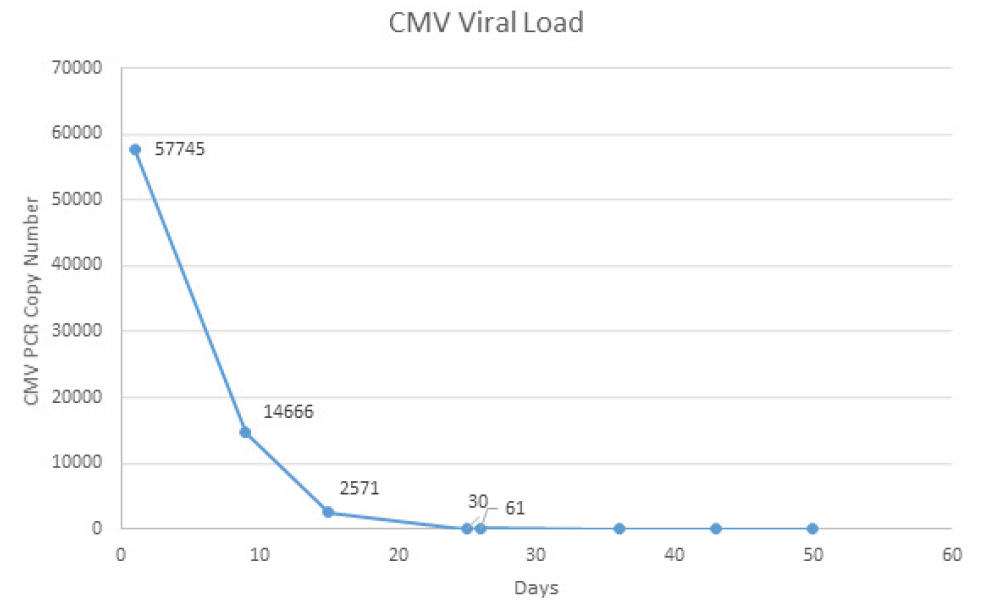
During his admission, the patient developed an acute respiratory failure requiring intensive care unit (ICU) admission and oxygen therapy with a high-flow nasal cannula. A chest CT showed multiple pseudo-nodular bilateral opacities, as well as bronchiectasis (Fig. 1). Meanwhile, once admitted to the ICU, a bronchoscopy was performed where CMV was detected by polymerase chain reaction (PCR) on the bronchoalveolar lavage (BAL), and isolation of Branhamella catarrhalis was made by positive culture. No malignant cells were observed. CMV viremia was also demonstrated, observing a blood count of 57,745 copies/ml (Fig. 2) and a positive IgM and IgG 325.00 U/ml serological test (positive ≥1.0 U/ml).
(click to enlarge)
Figure 1. A, B) CT scan showing cystic and cylindrical bronchiectasis in the middle lobe, lingula of the left lung and probably in the left lower lobe; C) Bilateral pseudo-nodular opacities with random distribution.
(click to enlarge)
Figure 2. CMV PCR copy number evolution of blood samples during ganciclovir treatment.
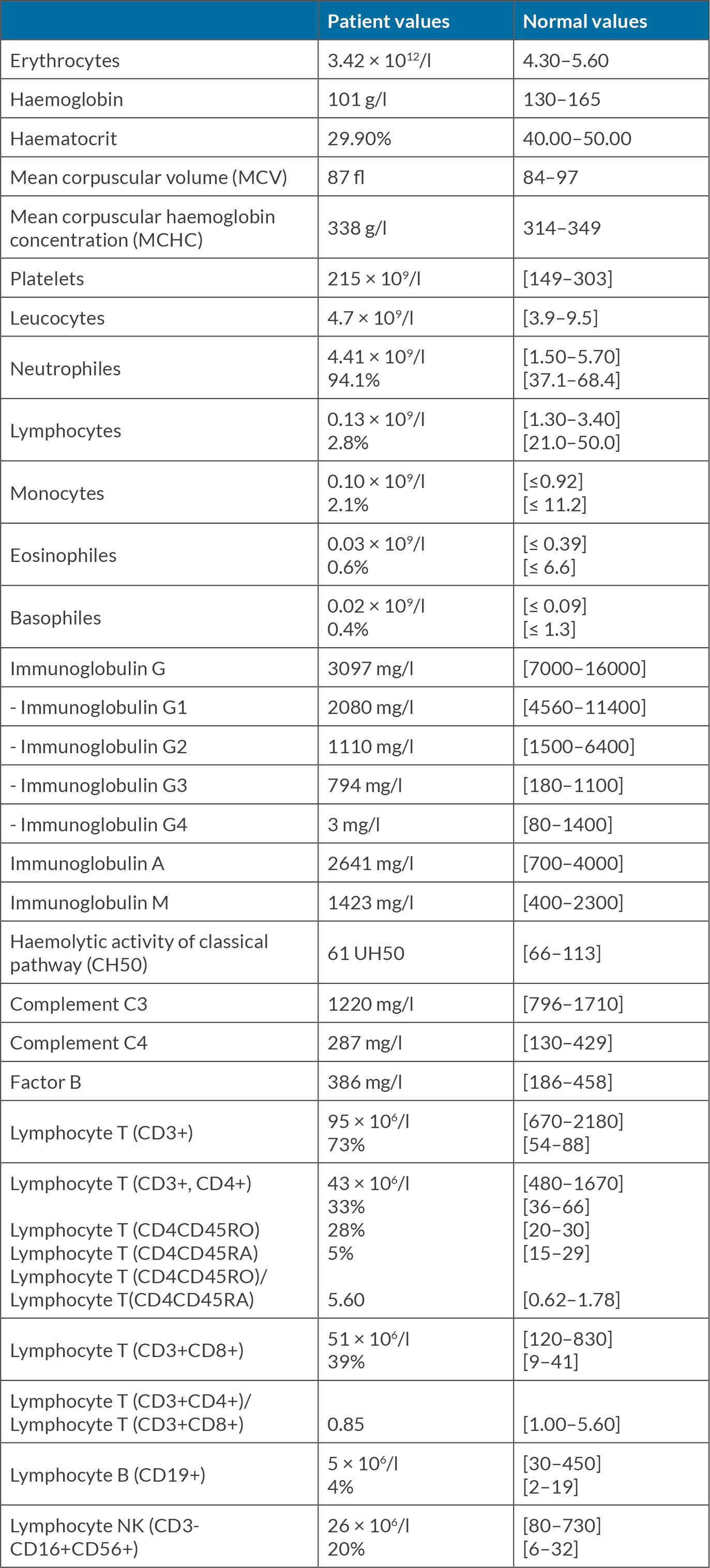
Interestingly, without receiving previous immunosuppressive treatments, the patient had exhibited grade 4 lymphopaenia in blood tests since his admission. Taking this into consideration, other OI were ruled out. HIV infection was discarded by two negative serological tests and a negative reverse transcription PCR test. We performed an immunological assessment (Table 1) that evidenced severe IgG deficiency affecting IgG1, IgG2 and IgG4 subclasses, with normal IgA and IgM, and severe lymphopaenia affecting all lymphocyte subtypes: T CD8+ lymphocytes and TCD4+ lymphocytes, natural killer lymphocytes (CD3-CD16+CD56+) and B lymphocytes (CD19+).
(click to enlarge)
Table 1. Blood tests and basic immunological assessment.
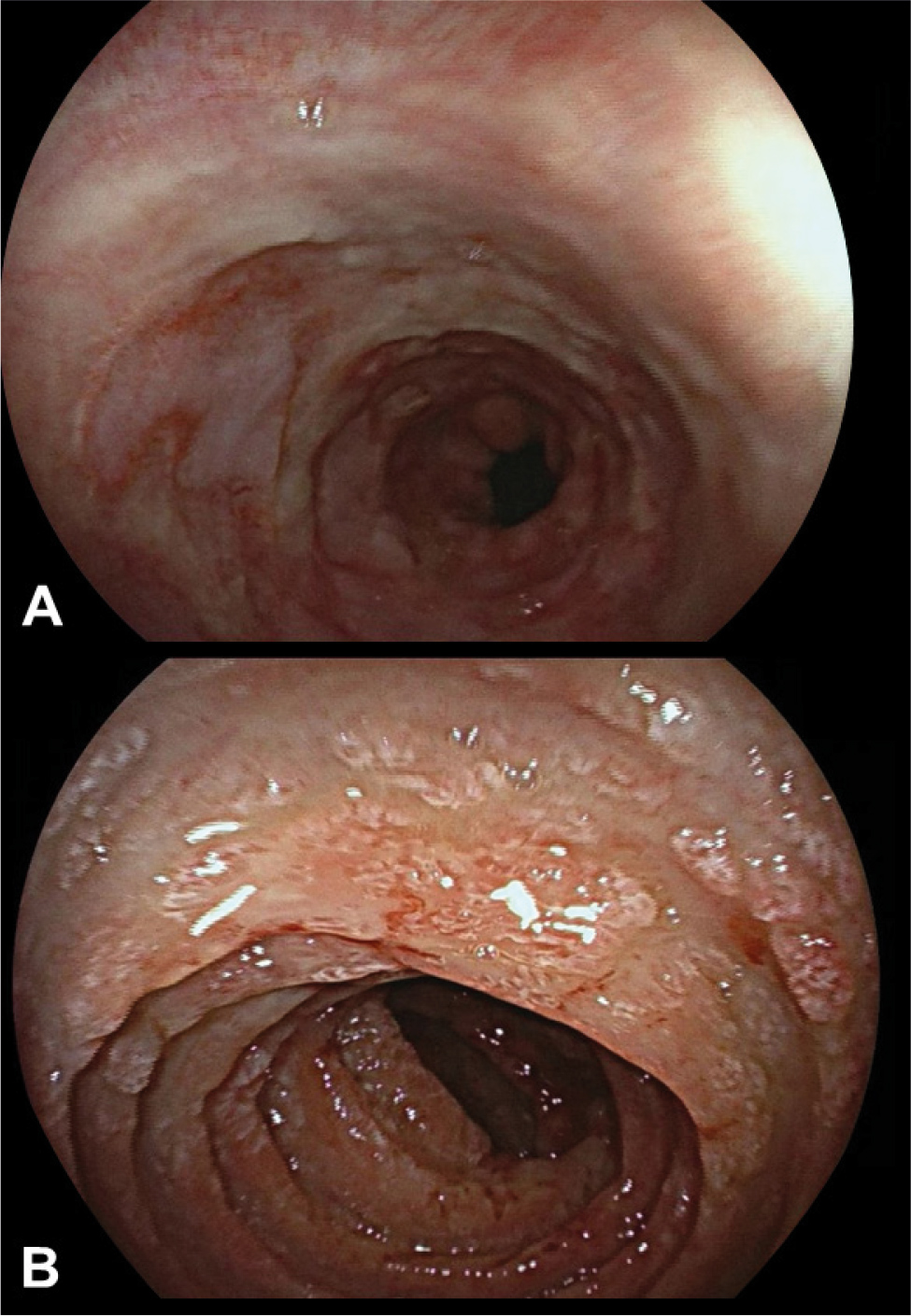
Antibiotic treatment with piperacillin and tazobactam, and antiviral treatment with ganciclovir 5 mg/kg IV twice a day was started, with initial favourable respiratory evolution. Subsequently, the patient developed a paralytic ileus with a CT abdominal scan showing diffuse thickening of the walls of the jejunum and distal bleeding. Due to the digestive involvement, an endoscopic study was performed (Fig. 3) showing diffuse affection of the oesophagus, ileum and colon. The anatomopathological analysis of the biopsies demonstrated inclusion bodies, positive immunohistochemistry stain for CMV and a positive PCR test.
(click to enlarge)
Figure 3. A) Oesophagogastroduodenoscopy evidencing serpentine geographical ulcers with a fibrin base. Also, erythematous veins, converging at multiple points; B) Jejunum with altered villous pattern, observing a friable, erythematous-oedematous mucosa with diffuse superficial ulcers.
The patient was diagnosed with a severe diffuse gastrointestinal CMV infection in a host with a subjacent LOCID. An FDG PET-CT scan was performed to rule out occult malignant diseases, which only revealed pulmonary and digestive hypercaptation, as well as multiple reactive mesenteric adenopathies. Prolonged antiviral treatment against CMV was prescribed. Given the severe T lymphocytopaenia, prophylactic treatment with cotrimoxazole was also started. Considering the severe IgG deficiency (<4.5 g/l), intravenous immunoglobulins were started at repositioning doses (0.4 g/kg every 3 weeks).
Despite the treatment, the patient had a poor evolution requiring re-admission to the ICU for persistent digestive bleeding after two-week hospitalisation in a general ward. In view of the refractoriness to antiviral treatment with ganciclovir, anti-CMV specific human immunoglobulin (Megalotect®) treatment was started. Despite all the therapeutic measures, the patient died from a refractory haemorrhagic shock. A necropsy was requested to confirm the diagnosis of CMV disseminated infection. The anatomopathological evaluation revealed the presence of acute ulcerative enteritis caused by CMV, involving the oesophagus, small intestine and sigma (Fig. 4), with the presence of active bleeding. Analysis of mesenteric adenopathies was not suggestive of lymphoproliferative syndrome, and no evidence of solid neoplasm was identified.
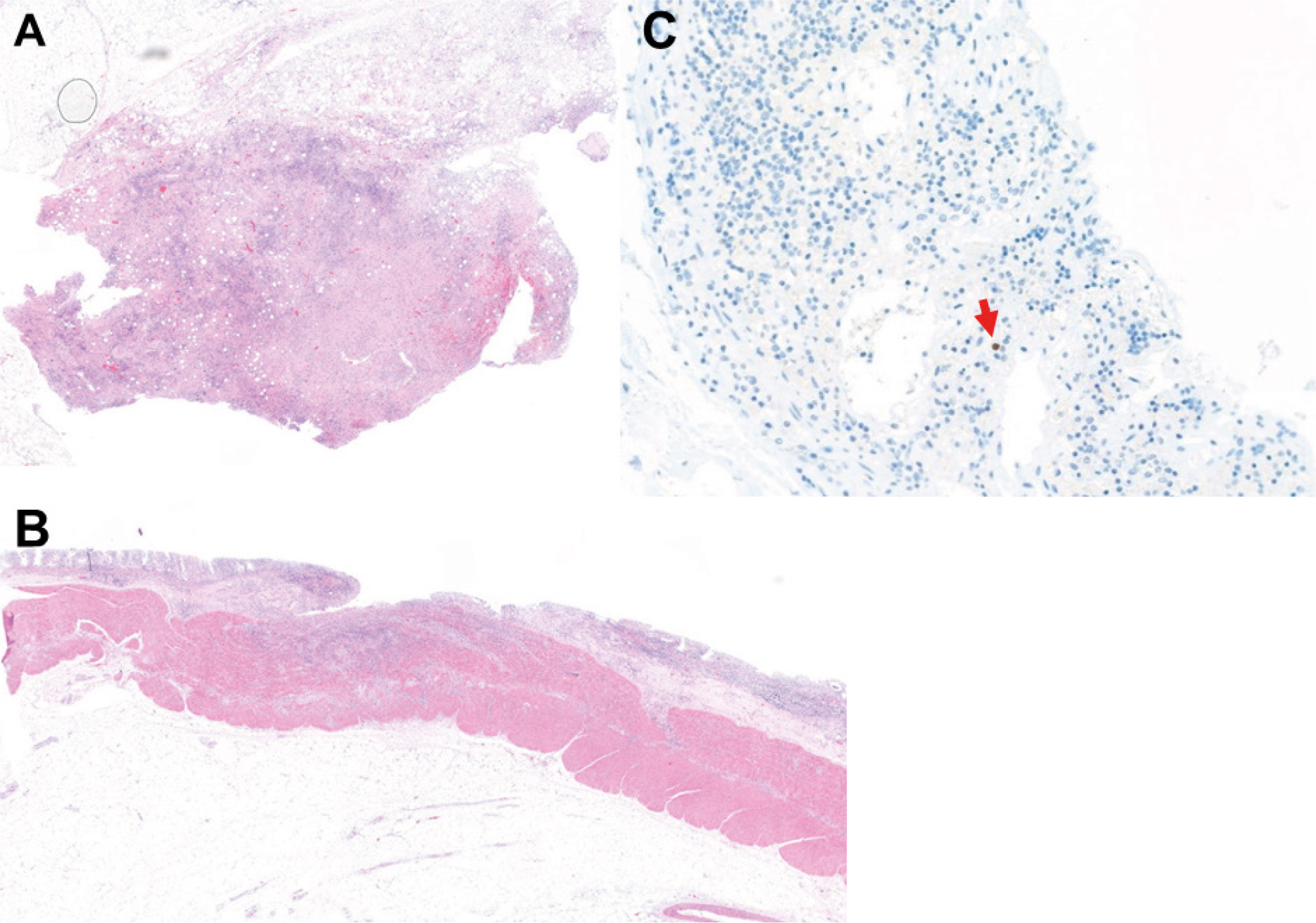
(click to enlarge)
Figure 4. Histology images of the necropsy. A) Small intestine showing an ulcer in the intestinal mucosa associated with transmural inflammation; B) Mesenteric lymph node showing predominantly histiocytic proliferation without malignancy; C) Monoclonal antibody against CMV antigen showed strong but focal CMV immunoreactivity with brownish area.
DISCUSSION
The diagnostic evaluation of an immunodeficient patient is always challenging. First, it is necessary to recognise the warning signs to identify a possible PID as, for example, recurrent and/or persistent or severe infections during childhood, infections with unusual or opportunistic microorganisms, inadequate response to antibiotics, persistent lymphopaenia and family history of PID. A prompt diagnosis will ensure the establishment of preventive measures and early treatment in case of complication. The diagnostic delay is associated with an increase in morbidity and mortality[1]. In this case, there was evidence that the patient suffered from an immunodeficiency: recurrent LRTI leading to bronchiectasis, PM and a diffuse and severe gastrointestinal CMV infection. Regarding the CMV infection, we also want to emphasise that CMV enteritis in immunocompetent individuals usually does not need a specific treatment, whereas it is recommended in immunocompromised hosts. In refractory cases, foscarnet, cidofovir and maribavir can be a useful option[7]. Also, as in our patient’s case, anti-CMV immunoglobulin (Megalotect®) can be added to anti-CMV specific treatment to induce the CMV viremia remission[8]. In some reported cases, restoration of immunity by infusion of CMV-specific T cells (‘adoptive T cells’) had been helpful[7].
Considering the severe and life-threating infection and refractoriness to the treatment that our patient suffered, we had no doubt of his immunocompromised subjacent status. The difficulty lies in discerning between a secondary immunodeficiency (SID) and a PID. A viral SID was reasonably discarded with multiple HIV tests and HTLV-1 and 2 negative serology. The patient did not have proteinuria and was not taking or had taken any treatment that could justify the hypogammaglobulinaemia. A malignancy related SID was reasonably excluded with the autopsy and the mesenteric adenopathies were examined carefully, taking into account the FDG PET-CT findings without finding alterations suggestive of neoplasia. Our final diagnostic approach was that the patient had a PID, considering the history of LRTI since youth, the presence of bronchiectasis and the cutaneous affection (PM). Of note, PM is a dermatological condition that could be related to immunosuppression, but more specifically, immunocompromised patients develop certain types of porokeratosis: PM, disseminated superficial actinic porokeratosis or a mix of them[9].
We classified the PID as a LOCID as the patient fulfilled the diagnostic criteria. This entity, considered a variant of the CVID, affects adults who present with OI and/or a low CD4 cell count[6]. Of note, our patient had a CD4+ T cell recount of 43 × 106/l [NV 480–1670], and also presented an OI in the form of a severe gastrointestinal CMV infection. With respect to CVID, patients with LOCID have higher rates of familial consanguinity, splenomegaly, gastrointestinal involvement (most frequently by CMV infection), lymphocyte depletion (mainly CD4+) and lymphoproliferative syndrome[2]. There is a lot of uncertainty regarding this rare and poorly studied entity. One limitation of this report is that we were not able to perform a genetic test to discard an adult-onset monogenic inborn error of immunity (MIEI) due to the rapid deterioration of the patient. As Staels et al. show in their review, the use of next-generation sequencing (NGS) in patients with PID has led to a rising tendency in identification of adult-onset MIEI[10]. The adult-onset phenotype could be associated with hypomorphic variants, somatic variants, epigenetics and environment[10]. It is necessary to deepen the knowledge of the LOCID and this is why we recommend carrying out genetic NGS tests in this population. Probably the term LOCID should be reserved for those patients who met diagnostic criteria without finding an alternative genetic cause.