ABSTRACT
This case report presents a rare case of cardiac leiomyomatosis misdiagnosed initially as submassive pulmonary embolism in a 39-year-old woman. The patient presented with syncope and hypotension, leading to an initial diagnosis of submassive pulmonary embolism. However, further investigations revealed a right-sided heart mass on echocardiogram. Surgical intervention was carried out, and the patient’s condition was successfully managed. This case emphasizes the importance of considering rare cardiac tumours in the differential diagnosis of pulmonary embolism.
LEARNING POINTS
- Given the rarity and diagnostic challenges associated with cardiac leiomyomatosis, it is important to raise awareness of this condition among healthcare professionals.
- Histopathological examination remains the gold standard for confirming the diagnosis of cardiac leiomyomatosis.
- Early recognition and accurate diagnosis are essential for timely intervention and optimal outcome.
KEYWORDS
Type A aortic dissection, Bentall procedure, atypical symptoms
INTRODUCTION
Cardiac leiomyomatosis is an extremely rare condition characterized by the presence of smooth muscle tumours within the cardiac chambers. These tumours are usually extensions of intravenous leiomyoma (IVL), which is rare benign smooth muscle cell tumour. The tumour originates in the uterus and intrauterine venous system, and can extend into the right side of the heart and pulmonary arteries[1,2]. Cardiac leiomyomatosis is often misdiagnosed initially due to its rarity and similarity with other cardiac pathologies. Misdiagnosis can lead to delays in appropriate management and potential adverse outcomes for the patient.
Given the rarity and diagnostic challenges associated with cardiac leiomyomatosis, it is important to raise awareness of this condition among healthcare professionals. Early recognition and accurate diagnosis are essential for timely intervention and optimal outcome. This case report aims to contribute to the existing literature on cardiac leiomyomatosis, and to emphasize the importance of considering rare cardiac tumours in the differential diagnosis of pulmonary embolism.
CASE PRESENTATION
A previously healthy 39-year-old woman presented to a local hospital after experiencing syncope. On arrival, her systolic blood pressure was 70 mmHg, but her haemodynamics quickly normalized. An initial CT scan suggested submassive pulmonary embolism, with a large volume of thrombus extending from the right ventricle to the right main pulmonary artery. Consequently, the patient was started on intravenous anticoagulation and considered for thrombolysis. However, further investigations, including an echocardiogram, revealed the presence of a right-sided heart mass.
On physical examination, the patient appeared comfortable with a temperature of 36.7°C (oral), heart rate of 73 beats per minute (peripheral), respiratory rate of 16 breaths per minute, blood pressure of 105/63 mmHg, and oxygen saturation of 99%. A cardiac examination revealed a systolic ejection murmur heard at the base of the heart and a right-sided.
SURGICAL INTERVENTION AND MANAGEMENT
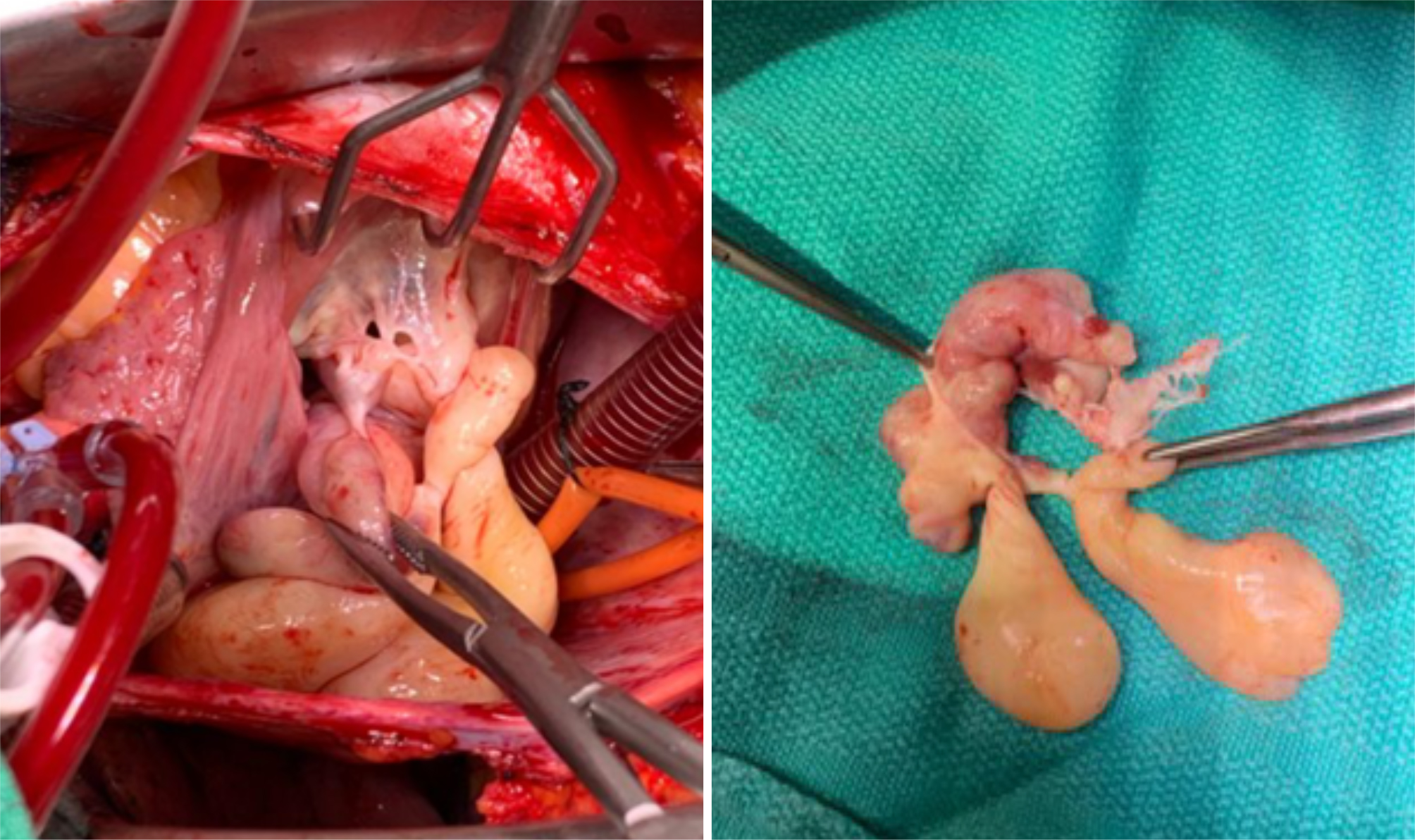
The patient underwent surgical intervention. Intraoperative images revealed an encapsulated tumour arising from the papillary muscle with the involvement of the anterior tricuspid valve leaflet (Fig. 1). A pulmonary artery incision was made, but no tumour or thrombus was found. Clear margins were achieved, and a 31 mm porcine valve was implanted during tricuspid valve replacement. The patient was placed on cardiopulmonary bypass for 57 minutes, with a cross-clamp time of 44 minutes. One chest tube and two right ventricular wires were placed without any complications.
(click to enlarge)
Figure 1. Operative view (left) and (right) tumours removed from the right atrium with the involvement of the anterior tricuspid valve leaflet.
The histopathological examination was consistent with multiple pedunculated, polypoid, lobulated, and worm-like tumours attached to valvular and chordal tissue. The tumour cells consisted of bland spindle-to-epithelioid cells with moderate to abundant eosinophilic cytoplasm. The tumours were diffusely and strongly positive for smooth muscle actin (SMA) and desmin.
Overall, the macroscopic, histomorphological, and immunohistochemical findings are most compatible with intracardiac leiomyomatosis.
DISCUSSION
Cardiac tumours, including leiomyomatosis, account for less than 0.03% of all cardiac neoplasms and are often challenging to diagnose and manage[2]. The symptoms and clinical presentation of cardiac leiomyomatosis can vary widely, ranging from asymptomatic cases to severe heart failure or sudden cardiac death[3]. Common presenting symptoms include palpitations, chest pain, dyspnoea, syncope, or signs of cardiac obstruction[4].
Diagnosing cardiac leiomyomatosis requires a high index of suspicion and the use of various imaging modalities. Echocardiography, cardiac MRI, and CT scans are commonly used to visualize the tumours and identify their location, size, and involvement of adjacent structures[5]. Histopathological examination remains the gold standard to confirm the diagnosis.
Surgical intervention is the main treatment for cardiac leiomyomatosis. The primary goal of surgical management is the complete resection of the tumour with clear margins[6]. In some cases where the tumour involves the cardiac valves, valve replacement may be necessary[7]. Long-term follow-up is crucial due to the potential for recurrence or metastasis.
CONCLUSION
We present a rare case of intracardiac leiomyomatosis misdiagnosed initially as submassive pulmonary embolism. This case highlights the importance of considering rare cardiac tumours in the differential diagnosis of pulmonary embolism, especially when initial management fails to provide the expected results. Early recognition and appropriate surgical intervention are crucial for optimal outcome.