ABSTRACT
While primarily described in children, adult-onset Langerhans cell histiocytosis (LCH) has been reported, albeit infrequently. In the present scenario, we unveil a unique case of adult-onset LCH in an HIV-infected individual. After the diagnosis was made, the patient was successfully treated and demonstrated total disease remission. This case illustrates the diagnostic challenge that rare clinical entities such as LCH pose, especially in the context of an untreated HIV infection. Furthermore, the complexity of treating adult-onset Langerhans cell histiocytosis in an HIV-positive patient is highlighted, with emphasis given on a multidisciplinary approach.
LEARNING POINTS
- Novelty: the case study provides knowledge on the uncommon occurrence of LCH in adults, especially within the setting of untreated HIV infection, underlining the significance of prompt detection and medical treatment.
- Diagnostic challenges: The scenario depicts the difficulty in diagnosing LCH in the presence of HIV, necessitating an array of diagnostic procedures.
- Multidisciplinary approach: This case’s effective management emphasises the crucial role of a multidisciplinary approach when dealing with complex medical conditions.
KEYWORDS
Langerhans cell histiocytosis, adult-onset, HIV
INTRODUCTION
Langerhans cell histiocytosis (LCH) is a systemic disorder defined by the accumulation of CD1a / Langerin-positive cells in a variety of tissues and organs[1]. Disease pathogenesis is based on the presence of activating somatic mutations in mitogen-activated protein kinase (MAPK) pathway genes, while inherited genetic variation is considered a determinant of LCH susceptibility[2]. LCH is a rare diagnosis, and it is considerably rarer in adults compared to the paediatric population. Accordingly, the existing literature is based on data from paediatric patients[1,3]. The annual incidence of LCH in adults is 0.07 cases per million per year for disseminated disease[3]. LCH can affect any organ, and the system involvement determines the clinical syndrome and severity of the disease. It can be easily misdiagnosed and its coexistence with HIV infection adds complexity to both diagnosis and management[1,4]. This report presents a unique instance of adult-onset LCH in an HIV-infected individual, emphasising the diagnostic challenges and the importance of a multidisciplinary approach.
CASE DESCRIPTION
A 33-year-old female patient from Kenya with a known medical history of HIV infection (CD4+ count of 70, viral load of 260,000 copies/ml) presented to the Infectious Diseases department with a two-month history of fevers, upper quadrant pain and fatigue. Despite a decade of known HIV seropositivity, the patient was non-adherent to antiretroviral therapy and was lost to follow-up. Physical examination revealed bilateral cervical lymphadenopathy, hepatomegaly and splenomegaly. Laboratory tests indicated cholestasis (total bilirubin: 5.9 mg/dl, direct bilirubin: 4.2 mg/dL, γ-GT: 494 IU/L, ALP: 1,310 IU/L) and anaemia (Hb: 7.6 g/dl, MCV: 75 fl). CT scans displayed cervical, mediastinal and abdominal lymphadenopathy. During these findings, suspicion arose of the existence of a haematologic malignancy in this adult patient with untreated HIV infection. To confirm such a diagnosis, a cervical lymph node biopsy was pursued. Histology was in accordance with the pathological description of LCH (Fig. 1). To create a comprehensive diagnostic picture and stage of the disease, further assessments were made. A bone marrow biopsy was conducted, followed by a skin biopsy, a brain MRI and the measurement of pituitary gland hormones. None of these investigations exhibited any signs of LCH infiltration. Finally, to comprehensively assess the extent of disease involvement and disease activity, a PET/CT scan was undertaken. The results displayed hypermetabolic lymph nodes, confirming active disease, and excluding other organ involvement (Fig. 2). Regarding treatment plans, after consultation with the Haematology department, a chemotherapy regimen was initiated, consisting of etoposide and dexamethasone. The patient received four monthly regimens in total, with etoposide titration depending on bilirubin values. In addition to LCH treatment, regarding the HIV infection the patient was started on antiretroviral therapy with tenofovir disoproxil, emtricitabine and dolutegravir (FTC/TDF + DTG). Subsequent laboratory and imaging monitoring demonstrated total disease remission and patient has been symptom-free for more than six months.
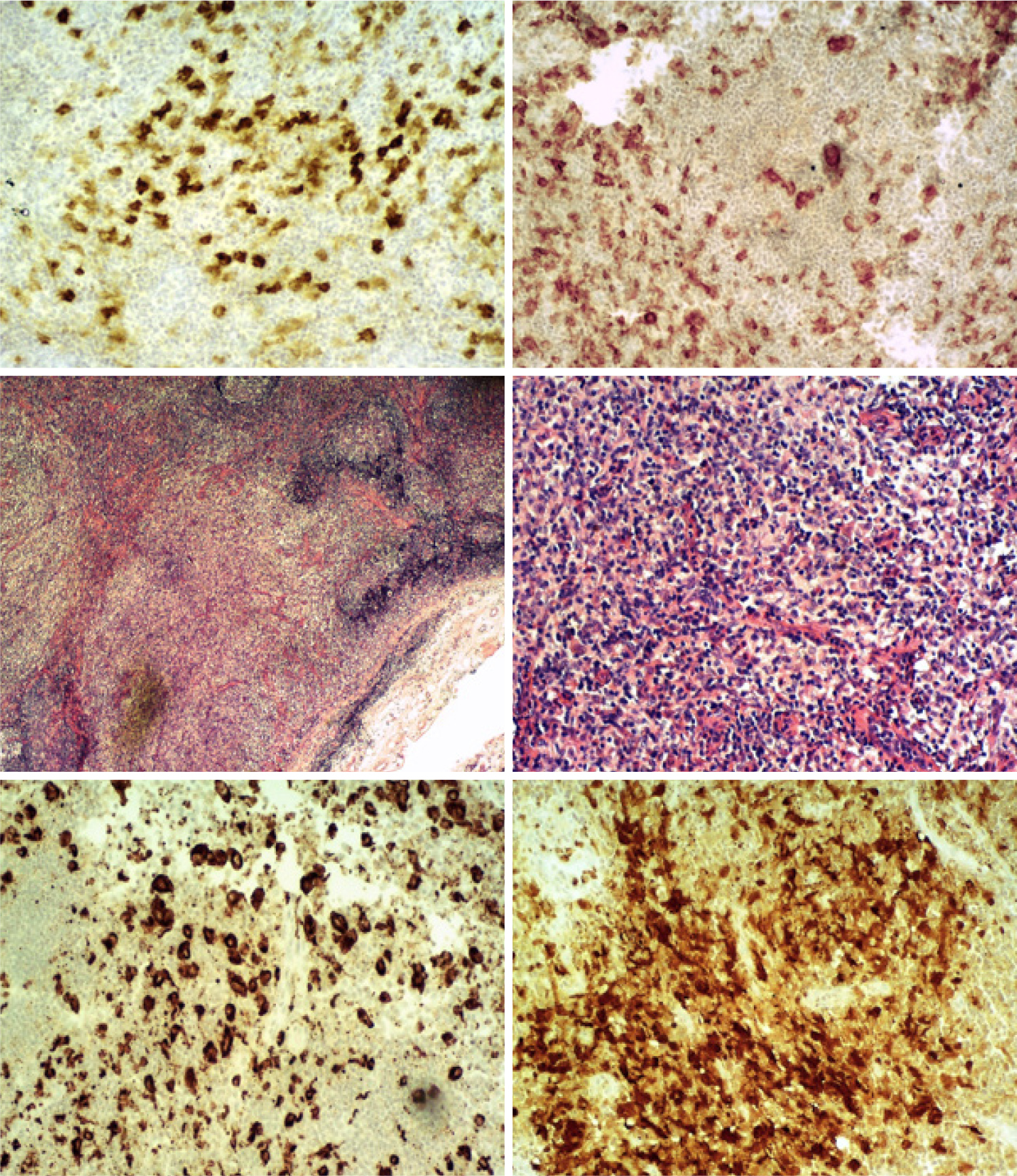
(click to enlarge)
Figure 1. Lymph node histology, from left to right: CD1a ×100, CD4 ×100, HE ×20, HE ×100, PGM1 ×100, S100 ×100.
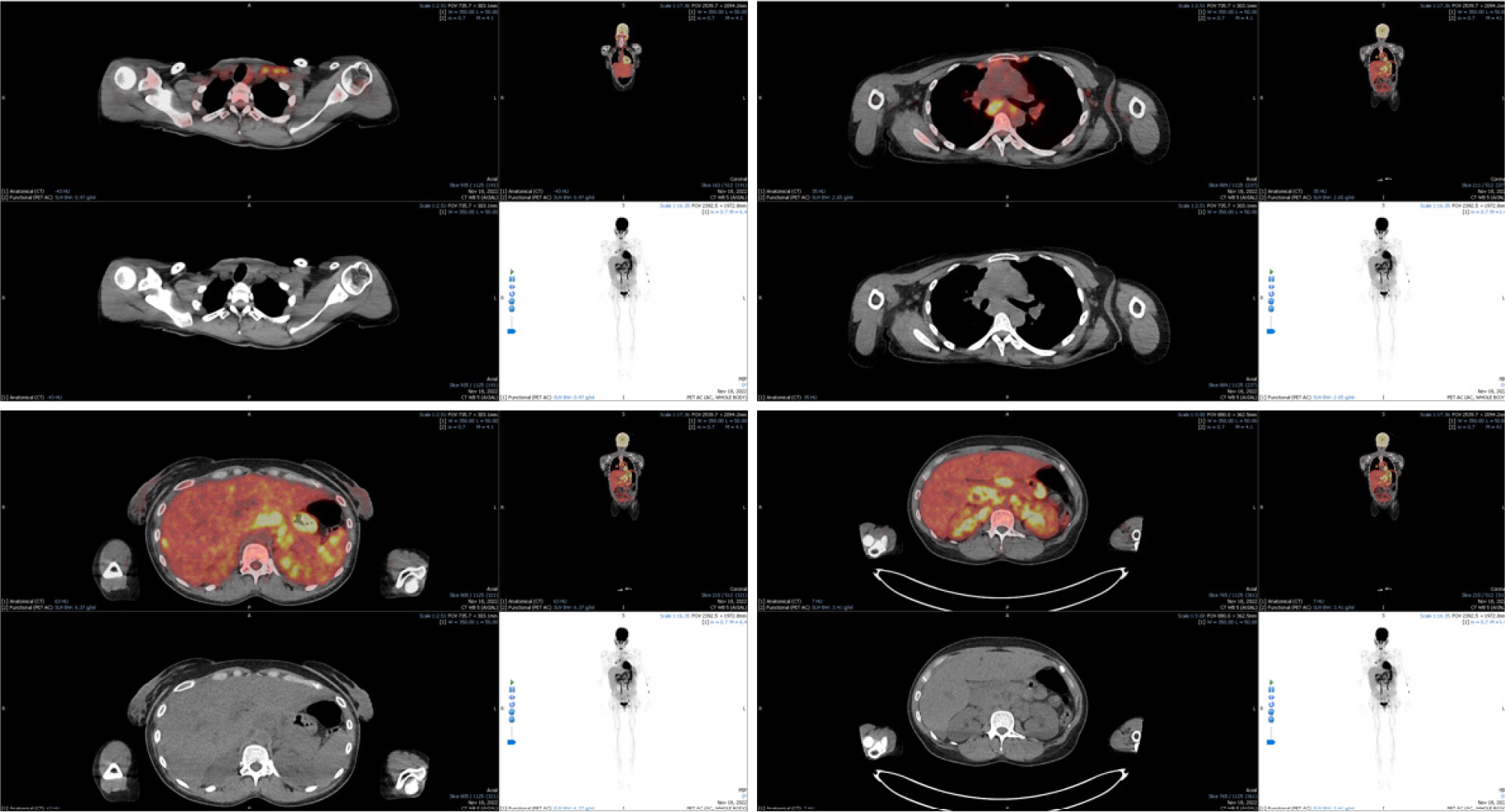
(click to enlarge)
Figure 2. PET/CT scan images showing hypermetabolic activity.
DISCUSSION
Langerhans cell histiocytosis is a histiocytic malignancy rarely affecting adults[5]. Owing to the establishment of clonality, LCH was one of the first histiocytic disorders to be acknowledged as a haematological neoplasm by the World Health Organization (WHO)[6]. LCH’s clinical presentation and natural course are exceedingly diverse, ranging from a benign, localised single-system disease which may reverse spontaneously, to various locations within one single-system disease, to a multisystem disease with life-threatening organ failure. The aspects of the disorder in adults continue to be insufficiently described[5]. Our case displays the diagnostic difficulties that may arise when LCH coexists with an HIV infection. The presence of LCH was concealed by common HIV-associated symptoms such as fevers, lymphadenopathy and hepatosplenomegaly.
Crucial to the diagnosis were the pathology findings of the cervical lymph node biopsy. The LCH infiltrate is characterised by the presence of clusters of intermediate-sized cells with reniform nuclei, frequent longitudinal nuclear grooves, dispersed chromatin and abundant eosinophilic cytoplasm. A reactive inflammatory cell background can also be also observed[6,7]. Our patient’s lymph node biopsy demonstrated a similar picture, with abundant histiocytes, lymphocytes and sporadic mature eosinophils. In addition, evident fibrosis separated the tissue structure in distinct nodules. Immunochemistry stains were positive for PGM1, CD1a, S 100 protein and CD4 on histiocytes. Many of them also stained using leucocyte common antigen (LCA) and CD56, and some with the CD15 stain (Fig. 1).
Adjunctively, a brain MRI with gadolinium and a focus on the sella turcica should be performed to exclude pituitary dysfunction. Bone marrow biopsy should be considered in the case of cytopenias or cytosis, due to the possibility of concomitant and subsequent myeloid malignancy in patients with LCH. A baseline full-body FDG-PET CT scan, including extremities, is recommended to facilitate diagnosis and determine the extent of the disease[5].
On the subject of treatment, multisystem LCH or extensive/refractory multifocal single-system LCH requires systemic chemotherapy with cladribine, cytarabine or vinblastine, prednisone[5,6]. In this HIV-infected patient, a chemotherapeutic regimen of etoposide and corticosteroids was chosen and was able to effectively control the disorder. Concomitant initiation of antiretroviral medication was essential for managing the underlying HIV infection.
CONCLUSION
LCH is a challenging diagnosis, and it should not be confined to the paediatric population. It requires a high degree of clinical suspicion, and reports of the disease in HIV-infected individuals are scarce. As illustrated in this case, prompt and thorough diagnostic assessment can result in effective treatment and remission, improving patient outcomes.