ABSTRACT
Wells syndrome or eosinophilic cellulitis is a rare and relapsing skin disease which lacks systemic involvement. A skin biopsy is needed to establish a diagnosis. Several precipitating factors have been proposed but no proven causative link has been found. On the other hand, Churg-Strauss syndrome, also known as eosinophilic granulomatosis with polyangiitis (EGPA), an auto-immune disease, is associated with multiorgan, including cutaneous manifestations. We report a case with overlapping features of Wells and Churg-Strauss syndrome, suggesting that these syndromes could be part of the same nosological entity.
LEARNING POINTS
- Biopsies are essential to establish a diagnosis in unusual cases of cellulitis.
- COVID-19 vaccination is a putative trigger for Wells syndrome.
- There is significant overlap between Wells and Churg-Strauss syndromes.
KEYWORDS
Wells syndrome, Churg-Strauss syndrome, eosinophilia, cellulitis, vasculitis
INTRODUCTION
Wells syndrome, first described in 1971[1], is a relatively rare condition with only 200 documented cases in the literature to date. Various triggering factors have been identified, including infections, insect bites, vaccines, drugs and malignancies[2].
The progression of Wells syndrome follows a distinct trajectory, with corresponding histological findings at each stage[3]. The initial stage is characterized by a burning sensation and the appearance of pruritic plaques, correlating histologically with eosinophil infiltration. Approximately seven days later, the syndrome transitions into the second stage, marked by tenderness, cutaneous oedema, and additional lesions; this is the phase where flame figures can be identified in histology. The resolution of lesions and the emergence of hyperpigmentation signify the third and final stage of the disease.
In terms of therapeutic options, prednisone is regarded as the standard of care. In corticosteroid-resistant cases, immunomodulatory drugs such as methotrexate[4], colchicine[5], bernalizumab[6], tacrolimus[7], dupilumab[8] and omalizumab[9] have been employed to manage the condition.
CASE DESCRIPTION
A sixty-nine-year-old man was admitted to a tertiary hospital with a three-week history of a mildly pruritic skin rash and progressive generalized oedema. Around the time of rash onset, the patient had complained of episodes of abdominal pain. Colonoscopy had resulted in unremarkable findings. Spontaneous remission of the rash had been followed by recurrence a few days later. He denied trauma, recent travel, illicit drug use or insect bites. His personal medical history was notable for bronchial asthma diagnosed after a COVID-19 infection 12 months prior to his presentation. He had been treated with bronchodilators since that time. Moreover, he had received the Pfizer-BioNTech vaccine against COVID-19 three months prior to admission. He had undergone a gastrectomy due to peptic ulcers and a cholecystectomy in his thirties. On clinical examination, there were confluent, annular, erythematous, maculopapular lesions with concomitant blisters throughout his body (Fig. 1) and generalized painless pitting oedema. He was afebrile and hemodynamically stable.
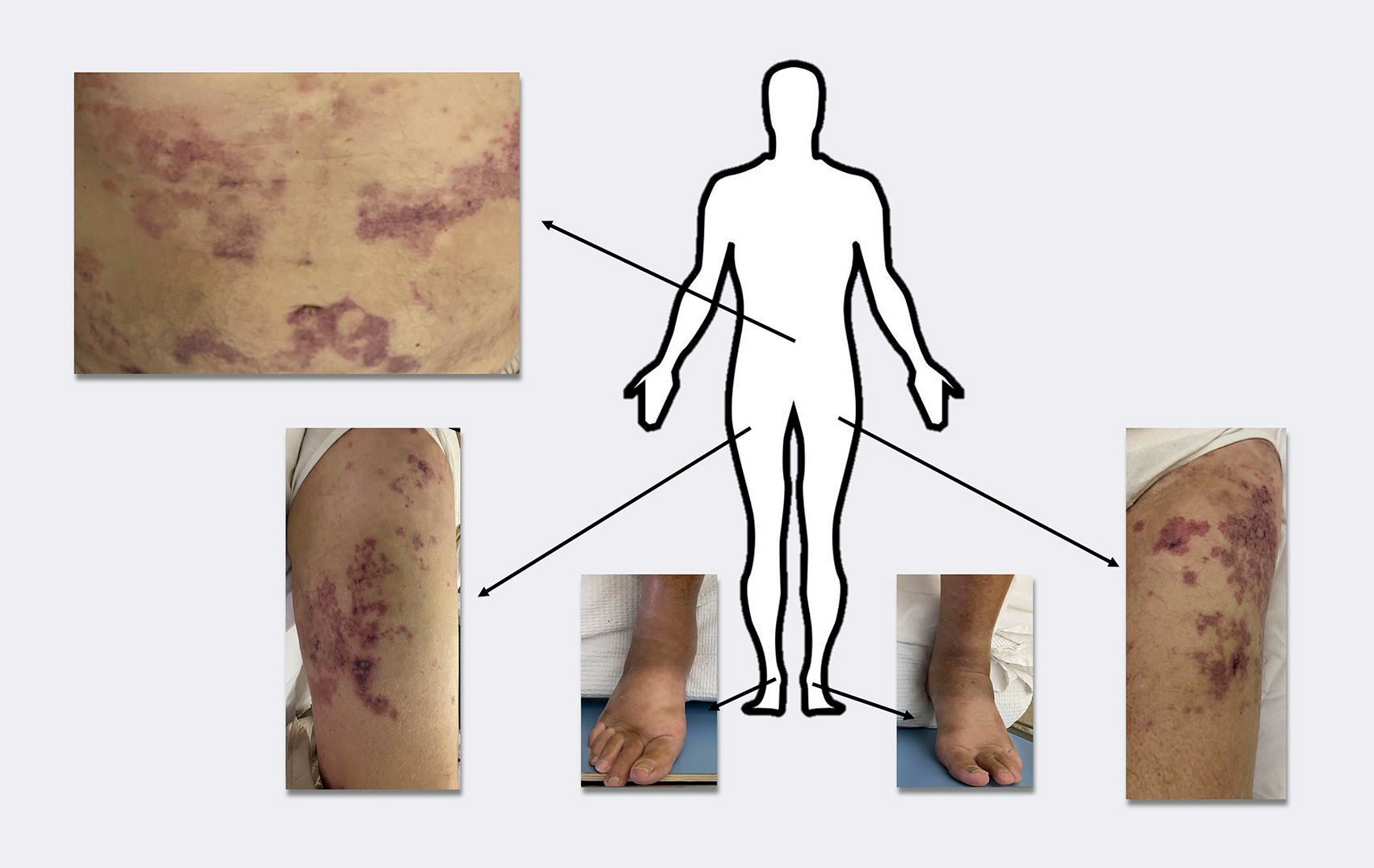
(click to enlarge)
Figure 1. Lesion distribution on the patient's body
Laboratory investigations revealed eosinophilia (24.4%, absolute eosinophilic number: 980/µl) along with hypoalbuminemia (2.5 g/dl) without proteinuria, and elevated levels of CRP (32.5 U/l, normal values: <6 U/l). Slightly decreased levels of C3 (50.5 mg/dl, normal values: 88-135mg/dl) and C4 (11.9 mg/dl, normal values: 16-27mg/dl) were also detected, while blood and urine cultures were negative, as were markers of collagen disorders including anti-neutrophil cytoplasmic antibodies (ANCA). His chest CT scan indicated a region of ground glass opacity in the right lung, whereas his head CT showed post-inflammatory lesions in the maxillary sinuses (Fig. 2). No abnormalities were detected on abdominal CT.
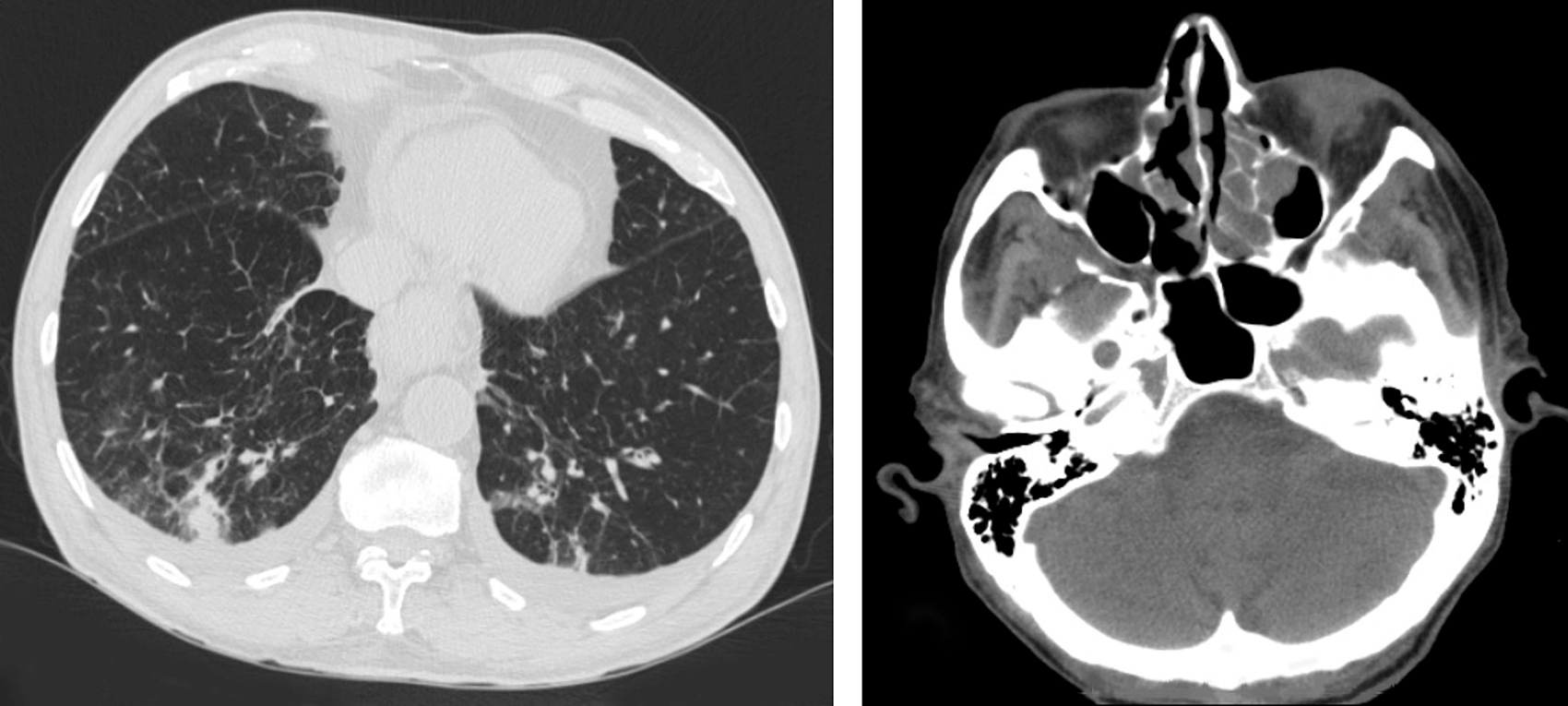
(click to enlarge)
Figure 2. Chest CT (left): Ground glass opacity in the right lung. Head CT (right): bilateral post-inflammatory lesions in the maxillary sinus
Several diagnoses were considered, some of which were excluded as the patient denied drug use, hypersensitivity reactions, and recent arthropod bites. The patient's physical examination was not consistent with the presence of compartment syndrome, necrotizing fasciitis or infectious cellulitis, and the imaging excluded any underlying solid-organ malignancies. The coexistence of eosinophilia, asthma and lung and paranasal sinus abnormalities raised the possibility of Churg-Strauss syndrome, in which cutaneous involvement has been reported in 40-70% of cases. A skin biopsy was deemed essential to establish a diagnosis. The histology demonstrated eosinonophilic infiltrates without signs of vasculitis, indicative of eosinophilic cellulitis (Fig. 3). This is a major criterion of Wells syndrome as delineated by Heelan et al[10], which together with the recurrence of the rash corroborated the diagnosis.
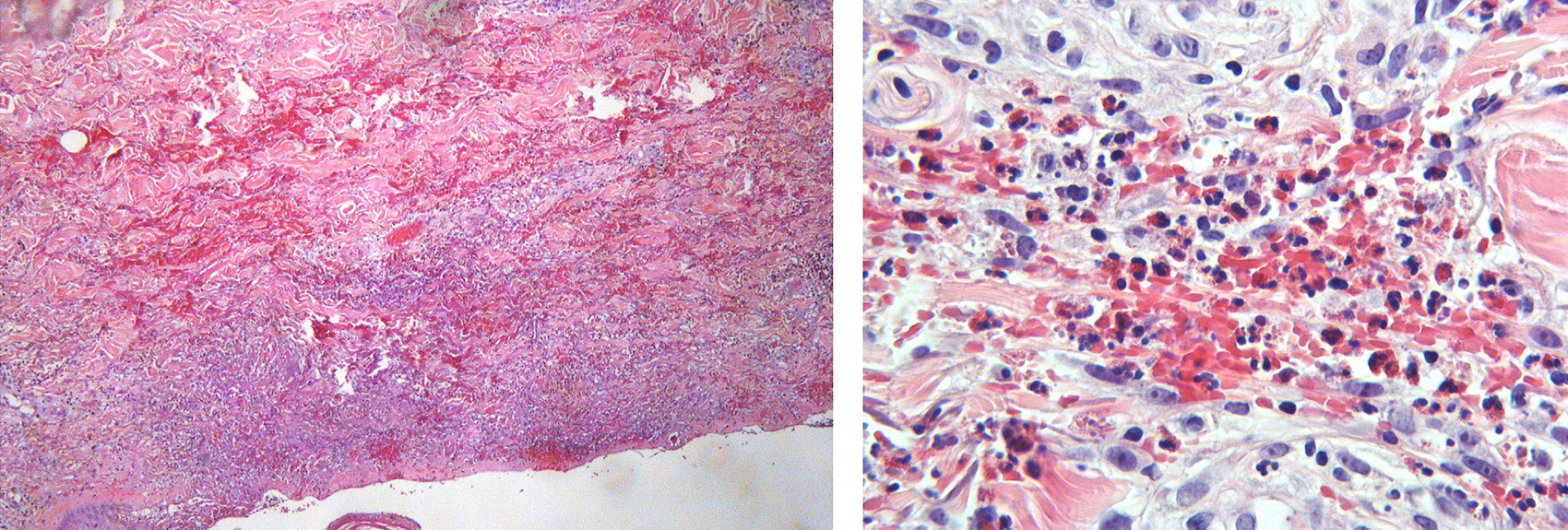
(click to enlarge)
Figure 3. Left: Extensive red cell extravasation and dense eosinophilic infiltration of the dermis with formation of subepidermal bulla (Haematoxylin and eosin stain x 100). Right: Large numbers of eosinophils intermingled with extravasated erythrocytes in the dermis (Haematoxylin and eosin stain x 400).
The therapeutic strategy was based on methylprednisolone, commencing with a daily dosage of 32 mg. It resulted in complete amelioration of the rash and oedema within two weeks, in parallel with normalization of all histological abnormalities. At the patient's monthly follow-up, the methylprednisolone dose was gradually reduced. One month post corticosteroid cessation, the patient presented with exacerbation of bronchitis without skin lesions, hinting at a dependency on corticosteroids. Nine months after his discharge, a skin rash reappeared and was treated with methylprednisolone. These instances of symptomatic flare-ups align well with the characteristically recurrent nature of Wells syndrome.
DISCUSSION
Systemic involvement is generally absent in Wells syndrome. The presence of certain features in this case - such as asthma, abdominal pain, oedema, hypoalbuminemia and abnormal paranasal and lung imaging - suggest the existence of a multisystem eosinophilic disorder. One should also consider the recent COVID-19 vaccination as a putative triggering factor. Notably, there are two published cases that link SARS-CoV-2 vaccination with the onset of Wells syndrome[11,12]. Moreover, in view of the observed low serum complement levels, we postulated that the oedema and hypoalbuminemia could potentially be attributed to a capillary leak triggered by immune complex deposition. To the best of our knowledge, however, there is no published evidence linking this phenomenon with Wells syndrome.
In light of the above, it was necessary to reconsider the diagnosis of Churg-Strauss syndrome. Despite the biopsy results negating the presence of vasculitis, certain considerations warranted further evaluation. Firstly, the selected biopsy site might have been inappropriate to effectively uncover vascular damage. Secondly, the evident eosinophilic infiltration raises the possibility that any vasculitis present might not have been distinctly identifiable. Lastly, the onset of vasculitis could potentially be a secondary development that had not yet manifested itself at the time of the biopsy.
While perinuclear anti-neutrophil cytoplasmic antibodies (p-ANCA) tend to be positive in Churg-Strauss syndrome, their absence in our patient's case does not exclude the diagnosis. The confirmation of vasculitis is not reliant on the presence of antibodies. There are recognized diagnostic criteria for Churg-Strauss syndrome including asthma, peripheral blood eosinophil count exceeding 10%, mononeuropathy or polyneuropathy, pulmonary infiltrates, paranasal sinus abnormality and extravascular eosinophils on biopsy. Notably, our patient satisfied four out of these six criteria[13]. A literature search identified twenty-six published cases[14–22] showing an overlap of Wells and Churg-Strauss syndromes. It is tempting to speculate that both syndromes are part of a single auto-immune disease.
CONCLUSION
In atypical presentations of cellulitis, while corticosteroids remain the gold standard to treat Wells syndrome, biopsies are indispensable to establish a differential diagnosis. Nevertheless, the substantial overlap observed between Wells and Churg-Strauss syndromes necessitates caution. This could potentially indicate that Wells syndrome may be just a reaction pattern rather than an independent entity.