ABSTRACT
Primary splenic lymphoma (PSL) is a rare disease and an improbable cause of splenomegaly or splenic nodules. On the contrary, splenic secondary involvement as part of an advanced lymphoproliferative disorder is more common.
The authors present the case of a 49-year-old woman with a primary splenic diffuse large B-cell lymphoma (PS-DLBCL), in which the absence of other organs’ involvement determined an ultrasound-guided biopsy of the spleen to achieve a definitive diagnosis.
With this case the authors intend to emphasise the extensive differential diagnosis of splenomegaly, splenic nodules or infiltrates, the usefulness of splenic biopsy in establishing the diagnosis and recall a rare disease, with non-specific presenting symptoms, in which the diagnostic workup is challenging.
LEARNING POINTS
- The differential diagnosis of splenic nodules or infiltrates is vast and challenging, and it includes haematological diseases, systemic infectious diseases but also non-malignant infiltrative diseases.
- Although some lymphomas frequently present with splenomegaly, this is not the case of DLBCL, with the exception of PS-DLBCL.
- PS-DLBCL is a very rare pathology, accounting for 1% of all DLBCL and less than 1% of all NHL.
KEYWORDS
Primary splenic lymphoma, diffuse large B-cell lymphoma, non-Hodgkin lymphoma, splenomegaly, splenic nodules
INTRODUCTION
PSL, characterised by the strict involvement of spleen and hilar lymph nodes, is a rare malignancy accounting for about 1% of all lymphoproliferative disorders[1,2]. Secondary involvement of the spleen as part of an advanced lymphoproliferative disorder is much more common.
The clinical presentation of non-Hodgkin lymphomas (NHL) varies with the histological subtype and sites of involvement. Some subtypes manifest variable lymphadenopathy for months or years, with or without constitutional symptoms, while others are aggressive enough to cause death within weeks, if left untreated. Even within a specific NHL subtype, the clinical presentation varies widely between patients, in a way that no manifestation is pathognomonic of a specific subtype, making NHL challenging to diagnose and unpredictable in its course. While diffuse large B-cell lymphoma (DLBCL) is the most frequent lymphoma accounting for 25% of all NHL[3], PS-DLBCL is uncommon, accounting for just 1% of all DLBCL.
CASE DESCRIPTION
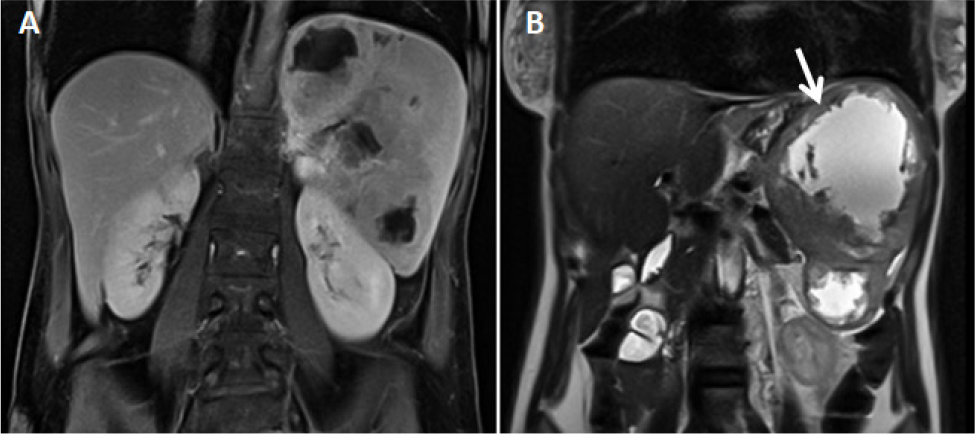
A 49-year-old woman, with no relevant personal or familial history, presented to the Emergency Department with upper left abdominal pain, early satiety, night sweats and an 8% weight loss within the previous two months. She reported no fever. On admission she was pale, tachycardic and with palpable splenomegaly, six centimetres below the left costal margin. Blood tests revealed a normochromic and normocytic anaemia (Hb 9 g/dl), thrombocytosis (445 x 109/l, reference range 150–400 x 109/l), LDH 892 UI/l (reference range 85–227 UI/l), erythrocyte sedimentation rate (ESR) of 101 mm/h, C-reactive protein (CRP) of 17.30 mg/dl (reference range < 0.60 mg/dl) and hypoalbuminemia (3.1 g/dl). The magnetic resonance imaging (MRI, Fig. 1) of the abdomen showed a multinodular splenomegaly of 15x11x17 (Fig. 1B, arrow).
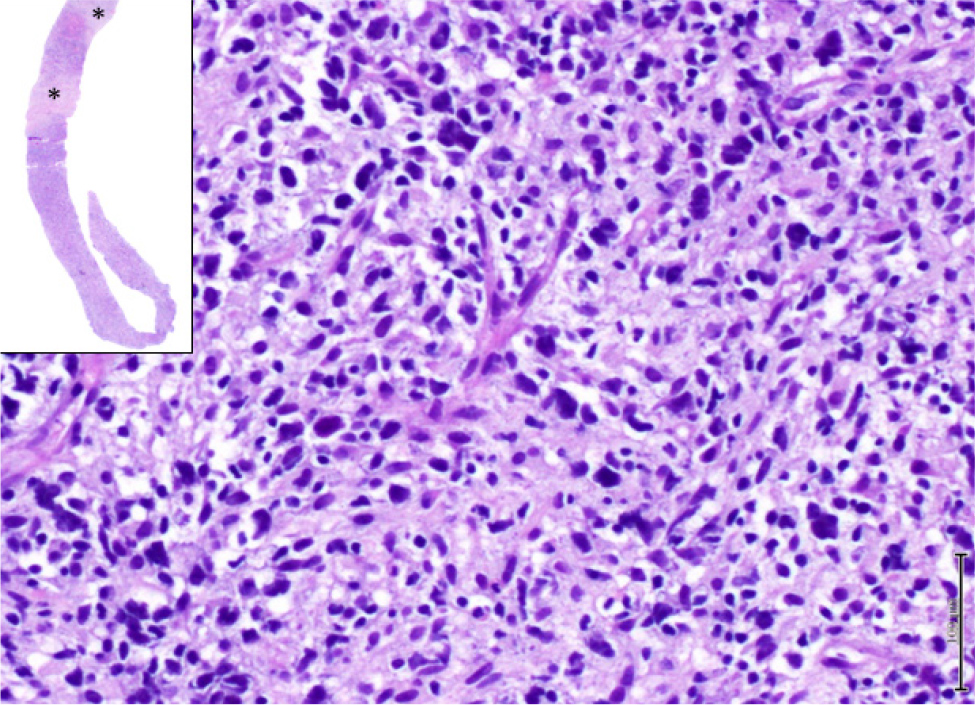
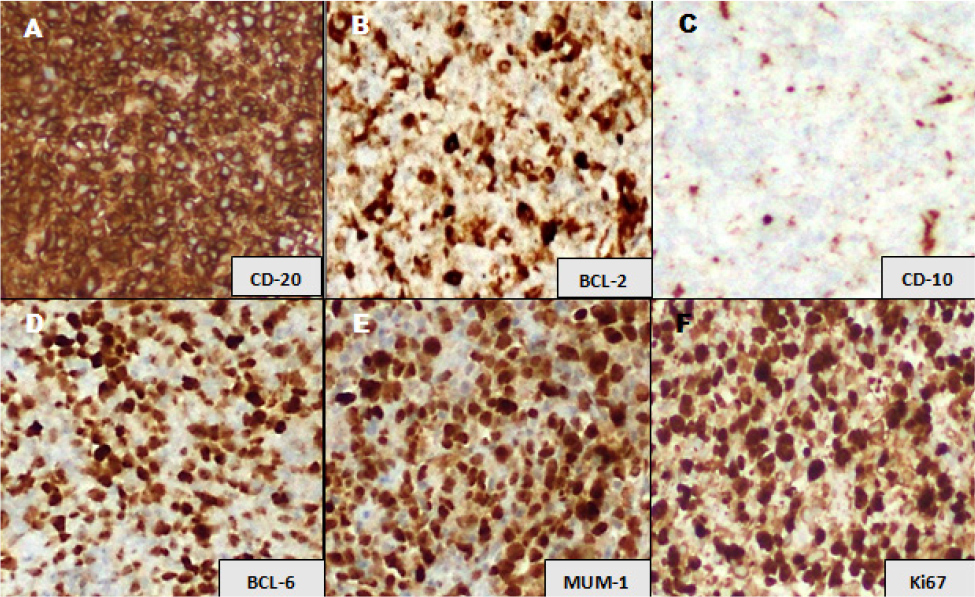
Serial blood cultures were negative as well as tests performed to exclude several infectious diseases such as infectious mononucleosis, cytomegalovirus, brucellosis, schistosomiasis, echinococcosis, human immunodeficiency virus, hepatitis B and C and tuberculosis. Myelogram and bone marrow biopsy showed no significant abnormalities – no leishmanias. There was no evidence of involvement of other organs, as confirmed by F-18 fluorodeoxyglucose positron emission tomography/computed tomography (18F-FDG PET/CT). An ultrasound-guided biopsy of the spleen confirmed a primary splenic DLBCL, with non-germinal centre B-cell-like (non-GCB) immunophenotype (Fig. 2 and 3), configuring a stage I disease. Her International Prognostic Index (IPI) score was 1 (low risk).
The patient started on first-line chemotherapy with conventional R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone) and experimental tafasitamab, a CD19-directed cytolytic antibody, approved for treatment of refractory, transplant-ineligible, DLBCL. At two years of follow-up the patient maintains complete remission.
(click to enlarge)
Figure 1. Abdominal MRI showing a non-specific multinodular splenomegaly, with multiple heterogeneous and confluent nodules, with cystic/necrotic expression, the largest measuring 11x9 cm (B, arrow). A: T1 weighted MRI, B: T2 weighted MRI
(click to enlarge)
Figure 2. Haematoxylin-eosin stained biopsy of the spleen showing areas of necrosis (*) and at higher magnification (x 400) revealing diffuse infiltration by neoplastic cells with large and hyperchromatic nuclei, with variably irregular nuclear contours and scant cytoplasm
(click to enlarge)
Figure 3. Immunophenotype featuring a non-germinal centre subtype diffuse large B-cell lymphoma. (A) Tumour cells are diffusely positive with CD20 immunohistochemical (IHC) stain; (B) positive with BCL2; (C) negative with CD10 IHC marker; (D) most tumour cells show immunopositivity for BCL-6; (E) strong nuclear expression is noted with MUM1 IHC marker; (F) Ki67 IHC stain depicts a high proliferative index (>90%) in tumour cells (x400 each)
DISCUSSION
DLBCL is the most common histological subtype of NHL, accounting for approximately 25% of all NHL cases[3]. Although the spleen can be considered as part of nodal tissue, splenomegaly is not typical of DLBCL. The most typical presentation is a rapidly enlarging symptomatic mass, usually lymph node enlargement, which can be found more frequently in the abdomen, neck or mediastinum (in cases of primary mediastinal DLBCL). The exception is PS-DLBCL, in which splenomegaly is the principal physical finding.
This is a rare disease, accounting for approximately 1% of all DLBCLs[3] and, consequently, less than 1% of NHL. There are other primary splenic lymphomas that can present with splenomegaly, such as splenic marginal zone lymphoma (the most frequent PSL), splenic red pulp small B-cell lymphoma and a splenic hairy cell leukaemia variant[3]. All PSLs are rare and their diagnosis is challenging, since histology of the spleen is mandatory and splenic biopsy is a risky procedure not readily available, but the only alternative to splenectomy.
However, since it is a rare disease, little is known about its presentation, prognosis and outcome. Bairey et al. studied 87 patients with PS-DLBCL aiming to evaluate presenting symptoms, prognostic factors and the diagnostic and therapeutic role of splenectomy in this lymphoma in the rituximab era. They found abdominal pain to be the most frequent symptom (81%) associated with PS-DLBCL, followed by B symptoms (59%)[3,4] including weight loss (51%), fever (24%) and night sweats (23%). These constitutional symptoms are non-specific to any particular condition and, although frequent in lymphoproliferative disorders, there are other non-malignant diseases that can present similarly (namely infectious diseases), requiring a high index of suspicion to not miss the diagnosis. Furthermore, they are not useful to distinguish between subtypes of lymphoma. Our patient presented with upper left abdominal pain and early satiety due to the enlarged spleen, as well as night sweats and a significant weight loss, which is consistent with the literature.
Characteristically, the spleen in this disease presents with one or more heterogeneous masses and/or nodules. In the above-mentioned study by Bairey et al., a splenic mass was detected in 97% of the patients at diagnosis, while 53% had multiple masses[4].
Independently of the presence of a splenic mass, the spleen was enlarged in 84% of the patients with a mean length of 17.3 cm[4], similar to our patient.
Regarding blood tests, LDH was elevated in 84% of the patients, CRP in 82% and hypoalbuminemia was found in 30%[4]. Thrombocytopenia less than 100x109/l was relatively rare and it was found only in 8%[4], despite the splenomegaly and hypersplenism usually associated with this lymphoma. Besides the expectable elevated LDH, CRP and ESR, our patient presented with an elevated platelet count, possibly due to the pro-inflammatory state caused by the disease.
Non-splenic DLBCL is often an advance disease at the time of diagnosis with an incidence of stage I disease around 28%, while this incidence is 42% for PS-DLBCL[3]. Accordingly, bone marrow and/or peripheral blood involvement in PS-DLBCL affects about 7–10% of patients, whereas in other PSL or lymphomas with secondary splenic involvement this is much more frequent[3,4].
Regarding stage I PS-DLBCL management, since it is a rare disease there are few data to guide physicians on the best therapeutic approach. Potential treatment modalities include combinations of splenectomy, radiotherapy and immunochemotherapy[5]. From this point of view, besides being the most effective mean of diagnosis, splenectomy can also be part of the treatment. However the complication rate of this surgery can be as high as 22% with a mortality rate of up to 4%[5]. Additionally, splenectomy imposes on the patient a state of susceptibility to infections due to encapsulated bacteria, which can delay the treatment.
The advent of rituximab in 2006 led to a decrease in the number of splenectomies, with therapeutic intention, without adversely affecting the prognosis of patients with PS-DLBCL[3,5]. This leads us to believe that any benefit resulting from splenectomy in this disease was drastically reduced after the introduction of this monoclonal antibody. This is corroborated by a retrospective analysis conducted by Byrd et al. in 2017, in which 470 patients with stage I PS-DLBCL were stratified by whether they were diagnosed before or after the regulatory approval of rituximab in 2006[3,5]. The authors found that after approval of rituximab, the rate of splenectomy decreased from 82% to 72% and the median overall survival time increased from 9 to 11 years[3,5]. Giving these facts, although splenectomy can still be necessary for diagnostic purposes, it is no longer mandatory for treatment.
Considering clinical presentation (stage I disease and an IPI of 1) the prognosis of our patient, with R-CHOP treatment, is favourable (3-year overall survival of 91% and 3-year progression-free survival of 87%)[6]. However, regarding molecular subtype, the non-GCB immunophenotype presented by our patient is associated with a worse prognosis following R-CHOP when compared to germinal centre B-cell-like (GCB) subtype. This was evidenced by Scott et al. in 2015 in a study including 344 patients treated with R-CHOP, which confirmed that the outcome at five years of patients with a non-GCB subtype DLBCL was inferior compared with patients with GCB DLBCL. This included freedom from progression (51%vs. 76%); progression-free survival (48% vs. 73%); disease-specific survival (61% vs. 82%) and overall survival (56% vs. 78%)[7].
Molecular studies have found MYC and BCL-2 double-hit lymphomas much more common among the GCB subtype, which are associated with inferior survival[8-10]. On the other hand, genetic aberration involving BCL-6 is associated with the non-GCB subtype but it does not seem to have an independent prognostic value[11]. In 2000, Capello et al. designed a study aiming to evaluate BCL-6 mutations throughout the spectrum of B-cell neoplasia, namely DLBCL. They concluded that BCL-6 mutations have a uniform distribution throughout the majority of DLBCL, with the exception of primary mediastinal DLBCL with sclerosis, suggesting a common origin from a germinal centre related B-cell. The overall survival of 72 patients with DLBCL, at a median follow-up of 68 months, was 58% in cases with a BCL-6 mutation and 44% in those without, with no significant difference between the two groups[12].
In our patient, after serologic exclusion of several infectious diseases and no diagnosis following an 18F-FDG PET/CT and bone marrow biopsy, a splenic ultrasound-guided biopsy was performed to distinguish between a lymphoproliferative disease, splenic abscesses or other infiltrative diseases.
It is important to ensure that splenic biopsies are performed by a skilled physician, in an experienced centre, minimising the possible complications inherent in the procedure and assuring that there is installed capacity to deal with complications in case they occur.
In our patient, the ultrasound-guided biopsy was essential to confirm the diagnosis, which highlights its importance in the context of splenic nodules or infiltrates. Splenic preservation allowed her to start immunochemotherapy earlier and with fewer complications arising from overwhelming post-splenectomy infections caused by encapsulated bacteria.