ABSTRACT
Introduction: Multicentric Castleman disease (MCD) is a lymphoproliferative disorder characterized by lymph node histopathology and systemic symptoms. To our knowledge, there are no descriptions in the literature of long-term outcomes of human herpesvirus-8 (HHV-8)-associated MCD.
Case Description: We report a case of a 70-year-old male living with human immunodeficiency virus and a history of human herpesvirus-8 (HHV-8)-associated MCD. The patient reported having had low-grade fever for two weeks. Extensive workup revealed systemic lymphadenopathy without evidence of autoimmune disease or malignancy. Lymph node biopsy was consistent with HHV-8-negative idiopathic MCD (iMCD). The patient was subsequently scheduled for anti-interleukin-6 therapy.
Discussion: The present case is the first report of probable development of iMCD after long-term follow-up for HHV-8-associated MCD. The case illustrates the possible long-term consequences of MCD, suggesting the necessity of further research on the pathogenesis of CD.
Conclusion: Given the uncertainty in the long-term outcomes of HHV-8-associated MCD, periodic surveillance of patients with a history of HHV-8-associated MCD is warranted. Prospective nationwide cohort studies comparing characteristics of HHV-8-associated MCD and iMCD would bring further insights.
LEARNING POINTS
- This is the first case describing the probable development of HHV-8-negative idiopathic MCD after HHV-8-associated MCD.
- Little is known of long-term outcomes of HHV-8-associated MCD and idiopathic MCD, necessitating periodic surveillance.
- HHV-8-negative idiopathic MCD patients are treated with siltuximab, an interleukin-6 inhibitor, unlike patients with HHV-8-associated MCD, who benefit most from rituximab.
KEYWORDS
multicentric Castleman disease, human immunodeficiency virus, interleukin 6, siltuximab
INTRODUCTION
Castleman disease (CD) is a rare lymphoproliferative disorder characterized by histological features such as lymphoid follicle hyperplasia and marked capillary proliferation with endothelial hyperplasia. The etiology and pathogenesis of CD remain unclear but could be related to chronic inflammation in the setting of other primary diseases, such as viral infections. Within CD, multicentric Castleman disease (MCD) is defined as CD associated with systemic lymphadenopathy and severe constitutional symptoms such as fever. MCD was initially described in 1983[1].
Although MCD was initially considered to occur in the sequence of human immunodeficiency virus and Kaposi sarcoma-associated herpesvirus/human herpesvirus 8 (KSHV/HHV-8) infections, it became clear in the 2010s that HHV-8-negative, idiopathic MCD comprises more than 50% of the total MCD cases. Clinically, symptoms of HHV-8-associated MCD and HHV-8 negative idiopathic MCD (iMCD) are similar, including constitutional symptoms such as fever and weight loss. iMCD has several clinical subtypes, including iMCD with thrombocytopenia, anemia, fever, renal dysfunction/reticulin fibrosis, and organomegaly (TAFRO) symptoms (iMCD-TAFRO); iMCD with idiopathic plasmacytic lymphadenopathy (iMCD-IPL); and iMCD without TAFRO or IPL features, classified as not otherwise specified (iMCD-NOS)[2,3]. Histologically, patients with HHV-8-associated MCD usually have plasma cell type pathology due to the integration of HHV-8 encoded viral interleukin-6 (IL-6) into plasmablasts. iMCD is histologically categorized into plasma cell and hypervascular types[4]. HHV-8-associated MCD is characterized by detecting HHV-8 either by latency-associated nuclear antigen-1 (LANA-1) in the lymph node or by polymerase chain reaction of peripheral blood.
While only limited data are available, the five-year overall survival of HHV-8-associated MCD with rituximab-based regimens is reported to be around 80–90%[5]. Approximately 15–20% of patients with HHV-8-associated MCD develop non-Hodgkin lymphoma. Given the rarity of the disease, however, long-term outcomes of HHV-8-associated MCD have not yet been reported in the literature. Here, we report on a case of iMCD thirteen years after the initial diagnosis of HHV-8-associated MCD.
CASE REPORT
A 70-year-old male with a medical history of HIV/acquired immunodeficiency syndrome (AIDS) on antiretroviral therapy and HHV-8-associated MCD diagnosed in 2010 presented to the emergency department with generalized weakness, exertional dyspnea, and low-grade fever. He reported experiencing the symptoms for two weeks.
He was initially treated for presumed pneumonia as an outpatient without improvement in his symptoms, which led him to visit the emergency department for further evaluation. He had been diagnosed with HIV/AIDS in December 2009 and started on treatment but had progressive fever and developed bilateral axillary lymphadenopathy in 2010. He subsequently underwent an excisional left axillary lymph node biopsy which showed prominent lymphoid follicles with prominent follicular dendritic cells, partially collagenized germinal centers, and blood vessels showing hyperplastic endothelium surrounded by thickened cuffs of collagen. With the positive HHV-8 immunohistochemical stains and no signs of malignancy, the pathologic diagnosis was highly indicative of HHV-8-associated hyaline vascular type CD. He was treated with rituximab and was in remission within a year. He has been adherent to all his medications, including his antiretroviral regimen.
On admission, he was noted to have a low-grade fever of more than 37.5 °C, and physical examination was notable for small and firm left cervical lymphadenopathy. The results of main laboratory testing are summarized in Table 1. They are notable for mild pancytopenia with hemoglobin 7.2 g/dl, platelet count of 105,000/µl, and mild acute kidney injury with a creatinine of 1.0 mg/dl. Extensive workup revealed normal lactate dehydrogenase, negative Coombs direct test, tuberculosis screening, cytomegalovirus deoxyribonucleic acid, and autoimmune disease panels. His HIV viral load was low at 434 copies/ml and he had a CD4 count of 358 x 106/l. His Parvovirus B-19 immunoglobulin M returned positive but was considered non-specific and unrelated to the current symptoms. The serum interleukin-6 (IL-6) level was elevated to 13.8 pg/ml. Computed tomography (CT) showed hepatosplenomegaly, enlarged mediastinal lymph nodes, and extensive retroperitoneal lymphadenopathy. Bone marrow biopsy showed 50–60% cellular marrow with erythroid hyperplasia and less than 5% blasts, with no granuloma, malignancy, or fibrosis. CT-guided retroperitoneal lymph node core needle biopsy showed lymphoid tissue with features suggestive of angiofollicular hyperplasia with negative HHV-8 immunohistochemical staining based on latency-associated nuclear antigen-1 or signs of malignancies, consistent with HHV8-negative iMCD. Hematology was consulted, and he was diagnosed with iMCD-NOS. The patient was discharged for outpatient management with plans to initiate siltuximab, an IL-6 inhibitor. During his outpatient course, while waiting for authorization for siltuximab, his condition improved spontaneously, and he is currently closely monitored to determine the need for siltuximab in the future.
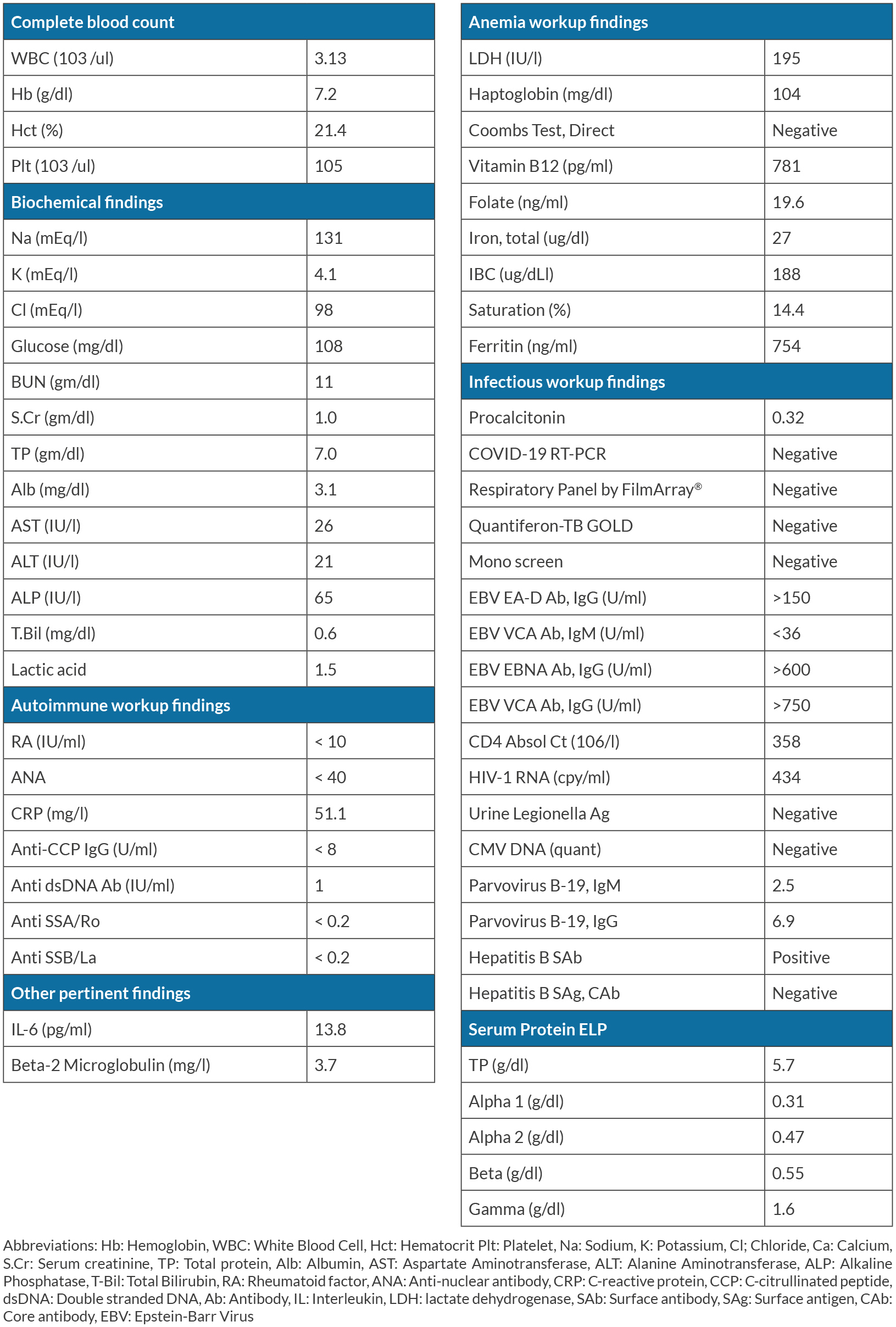
(click to enlarge)
Table 1. Main laboratory data
DISCUSSION
iMCD is a rare disease characterized by systemic symptoms and lymph node histopathology. To date, the relationship between HHV-8-associated MCD and HHV-8-negative iMCD has not been described. These diseases have been considered unrelated with distinct features. The present case is the first report of probable long-term development of iMCD-NOS in a patient with HHV-8-associated MCD.
As iMCD is more recognized by clinicians and pathologists, and better antiretroviral therapies have been developed for HIV/AIDS, the proportion of iMCD compared to HHV-8-associated MCD has been increasing. Relapse of HHV-8-associated MCD has previously been described in a cohort of 84 patients, and while HHV-8-associated lymphomas were reported during the five years of follow-up, no patient was diagnosed with iMCD during this time period[5]. Given that MCD could develop due to persistent chronic inflammation, immunodeficiency, or autoimmunity leading to overdrive of IL-6 production or sensitivity[6], patients living with HIV/AIDS, especially those in remission from HHV-8-associated MCD, regardless of HIV/AIDS control, could benefit from regular follow-up for early recognition of these lymphoproliferative disorders.
Treatment strategies for HHV-8-associated MCD and iMCD are different. The first line treatment for HHV-8-associated MCD is rituximab-based therapy, and it is critical to determine if there is concurrent Kaposi sarcoma to ascertain the need for chemotherapy.
For iMCD, anti-IL-6 therapy such as siltuximab and tocilizumab is the mainstay of treatment, depending on clinical subtypes and the extent of life-threatening organ failure. Currently, it is unclear if patients with higher serum IL-6 titers respond better to anti-IL-6 therapy, and if those with a history of HHV-8-associated MCD who now have iMCD may be better treated with re-introduction of rituximab instead of siltuximab. While nationwide cohorts for iMCD have been developed, a comparison of long-term outcomes and characteristics of HHV-8-associated MCD and iMCD is warranted.