ABSTRACT
Cystic fibrosis (CF) is a common autosomal recessive disorder which is mainly found in Caucasians but has also been reported in Asian populations. CF is primarily caused by mutations in the CFTR gene which regulates the transport of chloride ions across the cell membrane. We describe the cases of two siblings with CF diagnosed with the rare missense mutation c.80G>T, which has only been referenced once in the literature and shows a possible association with classical form of CF.
LEARNING POINTS
- c.80G>T is a very rare CFTR missense mutation which has not been known to be a disease-causing alteration.
- The mutation causes an amino acid switch from glycine to valine at position 27 in exon 2, resulting in the production of defective CFTR protein.
- In the homozygous state, c.80G>T seems to be associated with the classic CF phenotype.
KEYWORDS
Cystic fibrosis, Middle East, CFTR mutation, c.80G>T
INTRODUCTION
Cystic fibrosis (CF) is one of the most common autosomal recessive disorders among Caucasian populations. Since the identification of the cystic fibrosis transmembrane conductor regulator (CFTR) gene in 1989, more than 1800 molecular defects have been identified, ranging from point mutations, insertions and deletions to de novo alterations [1, 2]. Although a large number of mutations have been identified, a small handful account for most cases: in the European CF registry more than 80% of patients had at least one F508del mutation [3]. Mutation patterns in Middle Eastern populations are not clearly known but recent publications seem to show that F508del is not the most common mutation in this region and that a higher percentage of patients in this population have rare mutations [4]. Here we present the cases of two Emirati siblings (of Arab ethnicity) with a very rare CFTR mutation.
CASE 1
A 22-year-old man presented to the pulmonary clinic with long-standing productive cough and shortness of breath along with a history of chronic rhinosinusitis. He had never smoked and was married but infertile. He also reported recurrent admissions to hospital for chest infections since an early age, which were treated with antibiotics.
His treatments at presentation included nebulized Ventolin, budesonide, hypertonic saline and pancreatic enzyme replacement therapy. The patient reported he underwent daily chest physiotherapy in addition to recently starting long-term home oxygen therapy. On further questioning, he stated that he was given a probable diagnosis of CF 2 years previously on the basis of multiple positive sweat chloride tests; however, this was not confirmed with genetic analysis.
On examination, he had a low body weight with a BMI of 13, his oxygen saturation was 87–90% on room air, and he had a respiratory rate of 22 at rest, which he stated was his baseline. In addition, chest auscultation revealed bilateral coarse crackles.
He underwent extensive work-up at our hospital, including repeat sweat tests which were again positive. Faecal elastase testing confirmed pancreatic insufficiency. The initial test for 97 common CF mutations was negative. His pulmonary function test showed an FEV1 of only 0.63 litres (17% predicted). His DEXA scan T-score was –4. Lung imaging showed extensive, predominantly upper lobe bronchiectasis (Figs. 1 and 2).
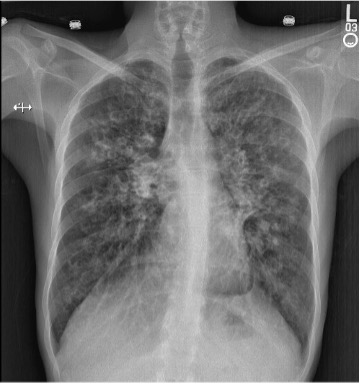
Figure 1 (click to enlarge)
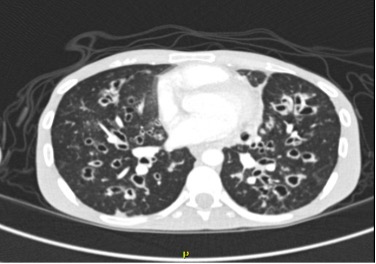
Figure 2 (click to enlarge)
Figure 1. The initial chest x-ray revealed hyperinflated lungs with diffuse extensive, fairly symmetric bilateral cystic bronchiectatic changes associated with bronchial wall thickening, suggestive of cystic fibrosis
Figure 2. CT of the chest without IV contrast revealed extensive bilateral bronchiectasis with marked worsening of the bronchial wall, a ground glass appearance and mosaic attenuation of the lungs suggestive of air trapping, and multiple areas of mucous plugging with an area of consolidation/atelectasis in the right upper and middle lobes
Because testing for common mutations was inconclusive, a CFTR DNA sequencing test was conducted, which showed the patient was homozygote for the c.80G>T mutation but did not reveal any previously known disease-causing CF mutations.
The patient was poorly compliant with treatment and did not attend the CF clinic for prolonged periods of time and hence continued to deteriorate. Although he was offered transplant assessment, he did not wish to consider a transplant. His clinical course was complicated by colonization by extended spectrum beta-lactamase Escherichia coli (ESBL E. coli) and methicillin-resistant Staphylococcus aureus (MRSA) in his sputum, causing recurrent exacerbations and worsening respiratory acidosis despite the use of nocturnal BiPAP, eventually requiring invasive mechanical ventilation. He died at age 27 from respiratory failure.
CASE 2
A 16-year-old girl, the younger sister of the patient described in Case 1, presented to the pulmonology clinic due to a long-standing history of productive cough with scanty yellow sputum and significant nasal congestion. The patient had a history of recurrent lower respiratory tract infections for which she usually received oral antibiotics. Her father reported that she had difficulty gaining weight and was on pancreatic enzymes for pancreatic insufficiency. She had been diagnosed with asthma and CF at the age of 10, but the family were not convinced of the diagnosis as they were told previous testing had not shown any CF mutations. On examination, the patient had a BMI of 14, respiratory rate of 21, and maintained 99% saturation on room air. Chest auscultation revealed crackles bilaterally, but there were no wheezes. Investigations revealed the FEV1 was 1.66 litres (64% predicted), she was negative for 97 common CF mutations, had low bone density on a DEXA scan and had pancreatic insufficiency. A CFTR gene sequencing test revealed she was a c.80G>T homozygote, the same missense mutation as her brother. The patient currently remains stable on treatment for CF.
DISCUSSION
We describe cases of two Emirati siblings with findings phenotypically consistent with classical CF and c.80G>T homozygosity. c.80G>T is a missense mutation which results in the substitution of glycine by valine at position 27 in exon 2. This is not clearly known to be a CF disease-causing mutation and is not mentioned in the comprehensive list of CFTR mutations maintained at the Cystic Fibrosis Mutation Database (CFMDB) (www.genet.sickkids.on.ca/cftr), which has 2110 mutations listed as of September 2022. However, this mutation has been reported previously in one study of Polish patients [5], and in 2016 a review of CF mutations in the UAE presented at the European Cystic Fibrosis Conference also described two patients with the c.80G>T mutation [6].
There is little literature regarding this mutation but given that the patients described here had the classic CF presentation with abnormal sweat tests, pancreatic insufficiency, predominantly upper lobe bronchiectasis and male infertility, it is reasonable to deduce that c.80G>T homozygosity causes the classic CF phenotype. Given that Panickar et al. [6] previously reported this mutation in paediatric patients in the UAE, it is likely that the prevalence of c.80G>T is higher in this region than so far documented.
In conclusion, c.80G>T is a rare mutation that in the homozygote state seems to be associated with the classic form of CF.