ABSTRACT
Introduction: Amyloidosis is a rare illness characterized by the deposition in organs of amyloid, which can be detected by histological staining. Amyloidosis restricted to the lower respiratory tract is unusual.
Results: We reported the case of a 68-year-old woman with no history of chronic disease who presented with dyspnoea on exertion, cough and fatigue. The physical examination was unremarkable. A CT scan showed a cystic mass with a thickened wall in the apical segment of the left lower lobe. A biopsy of the mass was performed, and histological and immunohistochemical study confirmed the diagnosis of AL amyloidosis. The patient’s clinical and radiological symptoms spontaneously improved without treatment after 3 years.
Conclusion: Amyloid-related cystic lung disease is a rare presentation of amyloidosis in the thorax. More case reports are required to determine if such masses can resolve without treatment and whether amyloid-associated cystic lung disease actually exists.
LEARNING POINTS
- Dyspnoea and cough are a rare atypical presentation that can reveal pulmonary amyloidosis.
- A cystic lung mass should raise suspicion for pulmonary amyloidosis.
- Clinical symptoms and radiological findings of a cystic mass spontaneously resolved without treatment after 3 years in this patient with pulmonary amyloidosis.
KEYWORDS
Amyloidosis, lung, pulmonary cystic mass
INTRODUCTION
Amyloidosis is a rare illness characterized by the deposition in organs of amyloid, which can be detected by histological staining [1]. The most common forms of amyloidosis are systemic AL amyloidosis (formerly primary amyloidosis), systemic AA amyloidosis (formerly secondary amyloidosis), systemic wild-type ATTR amyloidosis (formerly age-related or senile systemic amyloidosis), systemic hereditary ATTR amyloidosis (formerly familial amyloid polyneuropathy) and localized AL amyloidosis [2].The diagnosis is based on identification with Congo red stain and the subsequent demonstration of green dichroism under polarised light. The main organs involved are the kidney, heart, digestive tract, liver and skin [3]. Evolution is usually severe, with the destruction of the organs affected. Pulmonary amyloidosis is a rare disease and may be localized or occur together with systemic amyloidosis [4]. Amyloidosis restricted to the lower respiratory tract is unusual [5]. Here we report a rare case of pulmonary amyloidosis in the form of an isolated nodule suggestive of cystic mass.
CASE DESCRIPTION
A 68-year-old woman was admitted to our pulmonology department with complaints of worsening dyspnoea on exertion, cough and fatigue. The patient did not have a history of familial chronic disease. She was a non-smoker and had no pulmonary or systemic symptoms. The physical examination was unremarkable. Blood gases in ambient air showed respiratory alkalosis. Fibreoptic bronchoscopy did not reveal any endobronchial lesions, and bronchial fluid bacilloscopy was negative. A CT scan showed a cystic mass with a thickened wall in the apical segment of the left lower lobe (Fig. 1). Further laboratory tests showed the leukocyte count was 8,300/mm3, absolute neutrophil count 4,890/mm3, haemoglobin 13.7 g/dl and platelets 276,000/mm3. Biochemical analyses showed urea 4.7 mmol/l and creatinine 55 µmol/l. 24-Hour urine analysis revealed proteinuria 125 mg/24 h. The sedimentation rate was 12 mm/s and C-reactive protein was 0.45 mg/dl. The patient underwent a biopsy of the mass in the left lower lobe. Histologically, the mass was composed of a dense accumulation of eosinophilic, amorphous, homogeneous material, sharply demarcated from the surrounding lung tissue. Congo red staining produced an orange colour with apple-green birefringence under polarizing microscopy, features pathognomonic of amyloidosis (Fig. 2). The immunohistochemical study confirmed the diagnosis of AL amyloidosis (lambda light chain).
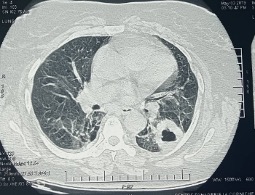
Figure 1 (click to enlarge)
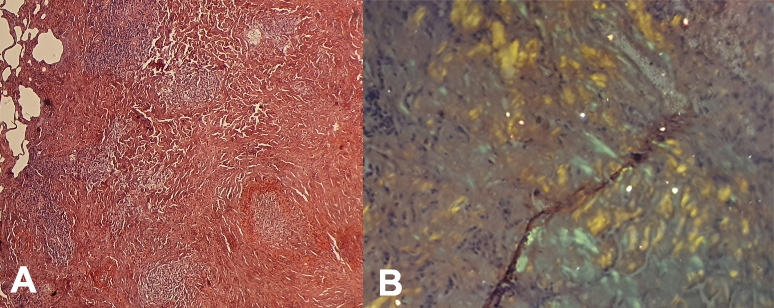
Figure 2 (click to enlarge)
Figure 1. CT image of the chest showing a cystic mass with a thickened wall in the apical segment of the left lower lobe
Figure 2. (A) Lung parenchyma with abundant amorphous interstitial deposits, coloured brick red by Congo red; (B) lung parenchyma under polarized light with apple-green birefringence confirming amyloidosis
A search for visceral and peripheral amyloidosis (macroglossia, peri-ocular lesion) was negative. The biopsy results of minor salivary glands were negative. A transthoracic echocardiogram was normal. Brain and cardiac MRI did not show any septal hypertrophy or signs of heart failure. Results of serum and urine electrophoreses were normal. A renal biopsy was not considered. An oesophageal-gastro-duodenal fibroscopy and a colonoscopy with staged biopsies were negative. After 3 years, the patient’s clinical symptom spontaneously improved without treatment, with resolution of the dyspnoea and cough and a reduction in the size of the cystic pulmonary mass.
DISCUSSION
Pulmonary amyloidosis most commonly presents as primary localized amyloidosis [6, 7]. Men are more usually affected than women, and the average age at presentation is 55–60 years [7]. Unlike systemic amyloidosis, localized pulmonary amyloidosis typically has a favourable evolution. The varied pattern of involvement of the lower respiratory tract results in several different clinical and radiologic presentations. Localized pulmonary amyloidosis may involve the tracheobronchial tree or pulmonary parenchyma with a nodular or diffuse distribution.
In pulmonary amyloidosis, amyloid deposits can either consist of amyloid light chain (AL) or be associated with amyloid A protein (AA), with AL deposition accounting for 63–80% of cases [8]. The literature on the distribution patterns of primary pulmonary amyloidosis is limited owing to its rarity.
Pulmonary amyloidosis localized without systemic deposition is uncommon, and is usually either tracheobronchial or parenchymal [5, 9]. Although some authors [8] have demonstrated that parenchymal nodular amyloidosis is more common than tracheobronchial amyloidosis, others [10] have shown the opposite.
Amyloid-related cystic lung disease is a rare. The mechanism of cyst formation in amyloidosis is not fully understood. However, one suggestion is that it is the result of air trapping within narrowed airways infiltrated by amyloid deposits and/or malignant lymphoid proliferation. Another hypothesis is that amyloid deposits within bronchioloalveolar structures and around alveolar capillaries could favour an ischaemic process leading to disruption of alveolar walls [11]. However, only a few case reports have been published so far. The diagnosis of isolated cystic pulmonary amyloidosis requires the absence of systemic amyloidosis, which was the case in our patient as all biopsies for systemic amyloidosis were negative. Cystic pulmonary amyloidosis is associated with benign or malignant lymphoproliferation, as well as connective tissue disease [4, 12, 13]. Our patient did not have pulmonary MALT lymphoma or a mixed connective tissue disease such as Sjögren's syndrome.
The novelty of our case lies in the localized pulmonary amyloidosis and the spontaneous resolution. The favourable clinical and radiological evolution with the disappearance of all symptoms, including dyspnoea and cough, was atypical. Although reports have shown that secondary amyloidosis can completely resolve with treatment, few cases of pulmonary amyloidosis have spontaneously resolved [14, 15]. To our knowledge, our case of pulmonary cystic amyloidosis is unique [14–16].
CONCLUSION
Amyloid cystic lung disease is rare. Imaging findings and patient symptoms are non-specific, underlining the importance of including amyloidosis in the differential diagnosis of cough and dyspnoea presenting together with a cystic lung mass. However, spontaneous resolution of this disease can occur without any specific treatment.