ABSTRACT
Pulmonary alveolar microlithiasis (PAM) is a genetic lung disorder that is characterized by the accumulation of calcium phosphate deposits in the alveolar spaces of the lung. PAM is discovered incidentally on radiographs performed for other purposes, and the typical disease course is characterized by slowly progressive respiratory failure over decades. Treatment remains supportive. A 62-year-old woman presented in the emergency department with dyspnoea and fatigue. On physical examination she had crackles on pulmonary auscultation and digital clubbing. A CT scan of the chest showed multiple high-density areas throughout the lung parenchyma, suggesting the presence of alveolar microlithiasis. This CT finding is the typical radiological presentation of PAM, while the hallmark presentation is clinical–radiological dissociation.
LEARNING POINTS
- Pulmonary alveolar microlithiasis (PAM) is a rare genetic lung disorder resulting in accumulation of calcium phosphate deposits in the alveoli.
- The typical radiological presentation of PAM is the classic ‘sandstorm’ appearance in the lung.
- The key to diagnosis of this disease is clinical-radiological dissociation.
KEYWORDS
Pulmonary alveolar microlithiasis, parenchymal calcification, clinical–radiological dissociation
INTRODUCTION
We describe a patient with pulmonary alveolar microlithiasis who presented with progressive dyspnoea and fatigue and was diagnosed based on typical imaging findings.
CASE DESCRIPTION
A 62-year-old woman, a non-smoker with a previous history of restrictive pulmonary pathology, was admitted in the emergency department with progressive dyspnoea and fatigue on medium effort. She denied fever, dry cough and chest pain. On physical examination, she was found to be asthenic and hypoxaemic with SpO2 of 82% on room air that improved to 94% on 3 l/min nasal O2. Bilateral crackles were noted on pulmonary auscultation together with finger clubbing. Routine biochemistry including serum calcium and phosphorus were normal. Chest radiography showed diffuse bilateral infiltrates (Fig. 1). A chest computed tomography (CT) scan showed multiple high-density areas throughout the lung parenchyma, suggesting the presence of alveolar microlithiasis (Fig. 2). These finding were deemed adequate to establish a diagnosis of pulmonary alveolar microlithiasis (PAM). The patient was discharged with supplemental oxygen and was referred for a lung transplant consultation.
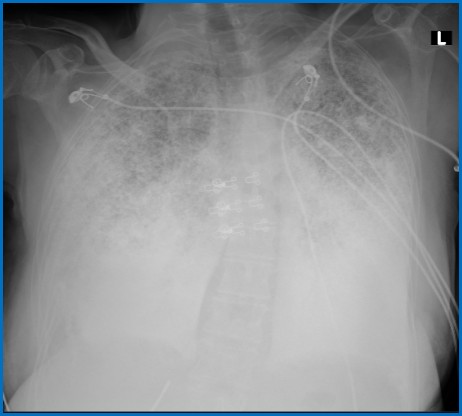
Figure 1 (click to enlarge)
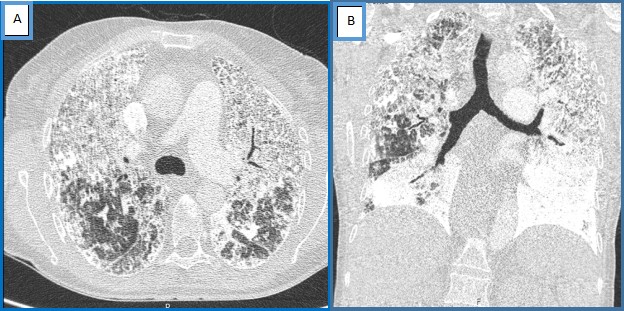
Figure 2 (click to enlarge)
Figure 1. Chest x-ray showing diffuse bilateral infiltrates
Figure 2. Chest CT scan showing bilateral diffuse intra-alveolar calcification: (A) axial view; (B) coronal view
DISCUSSION
PAM is a rare hereditary parenchymal lung disease characterized by the accumulation of calcium phosphate microliths in the alveoli [1-5]. A mutation in the SLC34A2 (solute carrier family 34 member 2) gene located on chromosome 4p15.2 produces a defective sodium-phosphate cotransporter in alveolar epithelial type 2 cells, making these cells unable to clear phosphorus released during recycling of surfactant [1-5]. Fewer than 1200 such cases have been reported globally [2, 3, 5]. This disease is typically diagnosed between 30 and 50 years of age [3, 4] and shows a slight female predominance, commonly in Asian and European populations [5].
Patients with PAM may initially be asymptomatic and are discovered incidentally after careful radiological examination for another medical purpose [2-5]. In symptomatic patients, shortness of breath is the most common symptom, as in the present case, followed by a dry cough, chest pain and asthenia [3,4]. Severe progressive disease may lead to respiratory failure [5].
A chest high-resolution CT scan shows numerous sandstorm-like intra-alveolar calcifications (microliths) throughout the lungs with pleural and peri-bronchial distribution, along with ground-glass opacities. The classic ‘sandstorm’ appearance with predominance in the middle and especially the lower lung areas, is the typical radiological presentation of PAM [3].
The key to diagnosis of this disease is clinical–radiological dissociation, as symptoms are less severe than suggested by imaging findings [2-5]. There is no known effective therapy and treatment remains mainly supportive [2, 3]. Long-term oxygen therapy is required for hypoxaemia and chronic respiratory failure [2, 4].
For patients with end-stage PAM disease, lung transplantation is the only definitive treatment [2, 3].
CONCLUSION
We describe a clinical case of PAM, which is a rare disease. Patients with PAM can be clinically asymptomatic or may present with non-specific signs and symptoms. A chest CT scan reveals bilateral sandstorm intra-alveolar calcifications. The disease cannot be cured, so treatment is supportive. Progressive disease may result in respiratory failure. Lung transplantation remains the only definitive treatment.