ABSTRACT
Acquired amegakaryocytic thrombocytopenia (AAMT) is a rare disorder of the bone marrow characterized by a lack of megakaryocytes and preservation of other cell lines. It can occur due to an intrinsic stem cell defect or secondary to viral infections, autoimmune disorders, lymphoproliferative disorders or environmental toxins. With time, it can progress to aplastic anaemia (AA) and can have a poor prognosis. No standard guidelines exist for the treatment of AAMT progressing to AA. Herein, we report a rare case of AAMT leading to AA and review the handful of cases previously published in the literature.
LEARNING POINTS
- Acquired amegakaryocytic thrombocytopenia can present as isolated severe thrombocytopenia which can initially be misdiagnosed as immune thrombocytopenia.
- Lack of response to steroids and intravenous immunoglobulin should raise suspicion for acquired amegakaryocytic thrombocytopenia.
- Over time, acquired amegakaryocytic thrombocytopenia can progress to aplastic anaemia, which confers a worse prognosis.
KEYWORDS
Acquired amegakaryocytic thrombocytopenia, aplastic anaemia, haematopoietic stem cell transplant
INTRODUCTION
Acquired amegakaryocytic thrombocytopenia (AAMT) is characterized by an isolated severe thrombocytopenia and an almost complete absence of megakaryocytes with preservation of other cell lineages. It is more commonly seen in middle-aged women. It is often misdiagnosed as immune thrombocytopenia (ITP) and can be an early presentation of aplastic anaemia (AA).
CASE DESCRIPTION
A 71-year-old woman presented in March 2020 with recurrent epistaxis, easy bruising, headaches and exertional dyspnoea of 1-month duration. She had smoked four cigarettes a day for 55 years and drank alcohol twice a week. Her family history was notable for lung cancer in her father. Physical examination revealed dried crusted blood at the nares and petechiae. Admission laboratory finding revealed a white blood cell count 3.2×103/μl, haemoglobin 5.5 g/dl, mean corpuscular volume (MCV) 111 fl, and platelet count 4×103/μl. Her reticulocyte count was 35.6×103/μl. Peripheral smear revealed macrocytic normochromic anaemia with mild anisocytosis, poikilocytosis and markedly reduced platelets. The iron panel, coagulation studies and haemolytic markers were normal. Her vitamin B12 levels were 200 pg/ml and serum folate 12.4 ng/ml. She received vitamin B12 supplements, packed red blood cell transfusions and platelet transfusions. Haematology recommended a computed tomography (CT) scan of the chest that was unrevealing.
Due to persistent headaches, a CT scan of the head was done which revealed a 9 mm acute subdural haematoma. Neurosurgery recommended no surgical intervention. A bone marrow biopsy showed substantially reduced megakaryocytes but otherwise a normocellular marrow, without dysplasia or abnormal cytogenetics, consistent with AAMT (Fig. 1a). A bone marrow differential revealed 50% cellularity, 5:1 myeloid-erythroid ratio, 2% blasts, 2% promyelocytes, 26% myelocytes, 15% metamyelocytes, 30% bands and neutrophils, 5% monocytes, 2% lymphocytes, 0% basophils, 1% eosinophils, 0% plasma cells and 16% erythroids. The patient’s anti-nuclear antibody (ANA), Coomb’s test, flow cytometry, hepatitis panel and HIV serology were unremarkable. After initial management, her haemoglobin stabilized; however, her platelet count remained low necessitating frequent transfusions. She was started on intravenous immunoglobulin (IVIG) and prednisone 1 mg/kg. Her repeat CT scan of the head showed a new 6 mm subdural haematoma. One week later, she had a second bone marrow biopsy, which showed the same findings as before (Fig. 1b). She was started on weekly rituximab 375 mg/m2 and prednisone was tapered off. After she failed to respond to three doses of rituximab, she was started on romiplostim 1 µg/kg/week. Her platelet counts transiently improved and she required platelet transfusions not more than three times a week. She was discharged home in April 2020 with close outpatient haematology follow-up.
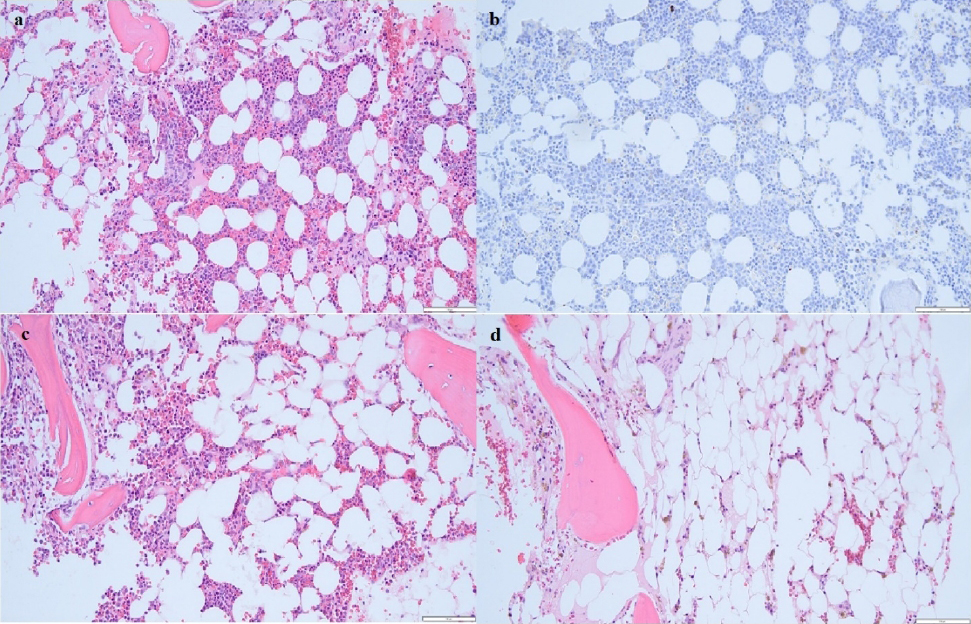
Figure 1 (click to enlarge)
Figure 1. (a) Haematoxylin and eosin stain of core bone marrow biopsy from March 2020 showing normocellular marrow with markedly absent megakaryocytes consistent with acquired megakaryocytic thrombocytopenia. (b) CD61 stain of core bone marrow biopsy from March 2020 revealing lack of megakaryocytes. (c) Haematoxylin and eosin stain of core bone marrow biopsy from May 2020 showing hypocellular marrow with no megakaryocytes. (d) Haematoxylin and eosin stain of core bone marrow biopsy from February 2021 showing less than 10% marrow cellularity consistent with aplastic anaemia.
After discharge, the patient received a fourth dose of rituximab and romiplostim was gradually increased to a maximum dose of 10 µg/kg/week to maintain platelet counts at more than 20×103/μl. Romiplostim was tapered off after a total of 13 weeks of treatment due to insufficient response. She had a third bone marrow biopsy in May 2020, which showed no megakaryocytes and mild dysplastic changes (Fig. 1c). Cytogenetics was normal and next generation sequencing was negative. Thereafter, she was started on cyclosporine 2 mg/kg two times daily with a target serum level of 150–400 ng/ml. She was continued on cyclosporine for 3 months but had no significant response. In August 2020, she was started on azathioprine 200 mg daily. She remained on azathioprine for about 1 month but it was stopped after she developed gingival bleeding and neutropenic fever. In October 2020, she declined a referral to a bone marrow transplant (BMT) centre. In February 2021, she had a fourth bone marrow biopsy for evaluation of worsening pancytopenia, which revealed less than 10% marrow cellularity, suggestive of aplastic anaemia (Fig. 1d). In March 2021, she was started on eltrombopag 50 mg daily that was slowly up titrated to 125 mg daily. She remained on it until September 2021 when she self-discontinued it. Her platelet count fluctuated around 30×103/μl and haemoglobin remained stable at 8 g/dl. The plan is to start anti-thymocyte globulin (ATG) with tacrolimus in the future.
DISCUSSION
AAMT can be due to an intrinsic stem cell defect or secondary to viral infections, autoimmune disorders, lymphoproliferative disorders or environmental toxins[1]. AAMT can eventually progress to either aplastic anaemia (AA) or myelodysplasia[2]. Currently, there are no standard treatment guidelines for AAMT progressing to AA[1]. Hoffman et al. proposed an algorithm for AAMT treatment, depending on aetiology[3]. AAMT with a known aetiology can be treated with withdrawal of the offending agent, plus steroids or occasionally even antiviral therapy. Those who have an anti-MK antibody can benefit from immune-suppressive treatment, and those with T-cell mediated inhibition of megakaryopoiesis can be treated with ATG with or without the addition of cyclosporine therapy. Levy et al. reported an AAMT patient who progressed to AA but was responsive to cyclosporine, ATG and eltrombopag[2]. Novotný et al. described a case of AAMT progressing to AA, which remained refractory to eltrombopag, romiplostim and cyclosporine and ultimately required work-up for a bone marrow transplant[4]. Simkins et al. described a patient with AAMT and pure red cell aplasia (PRCA) associated with thymoma, that ultimately progressed to refractory AA, who was treated with bone marrow transplantation as a last resort[5]. Our patient ultimately responded to eltrombopag with plans for ATG and tacrolimus at the time of last follow-up. Patients with refractory or relapsing AAMT can also be considered for myeloablative therapy followed by bone marrow transplantation[5]. Given the lack of established treatment guidelines for AAMT and its progression to AA, we aim to provide a consolidated review of all reported cases and its treatment options (Table 1).
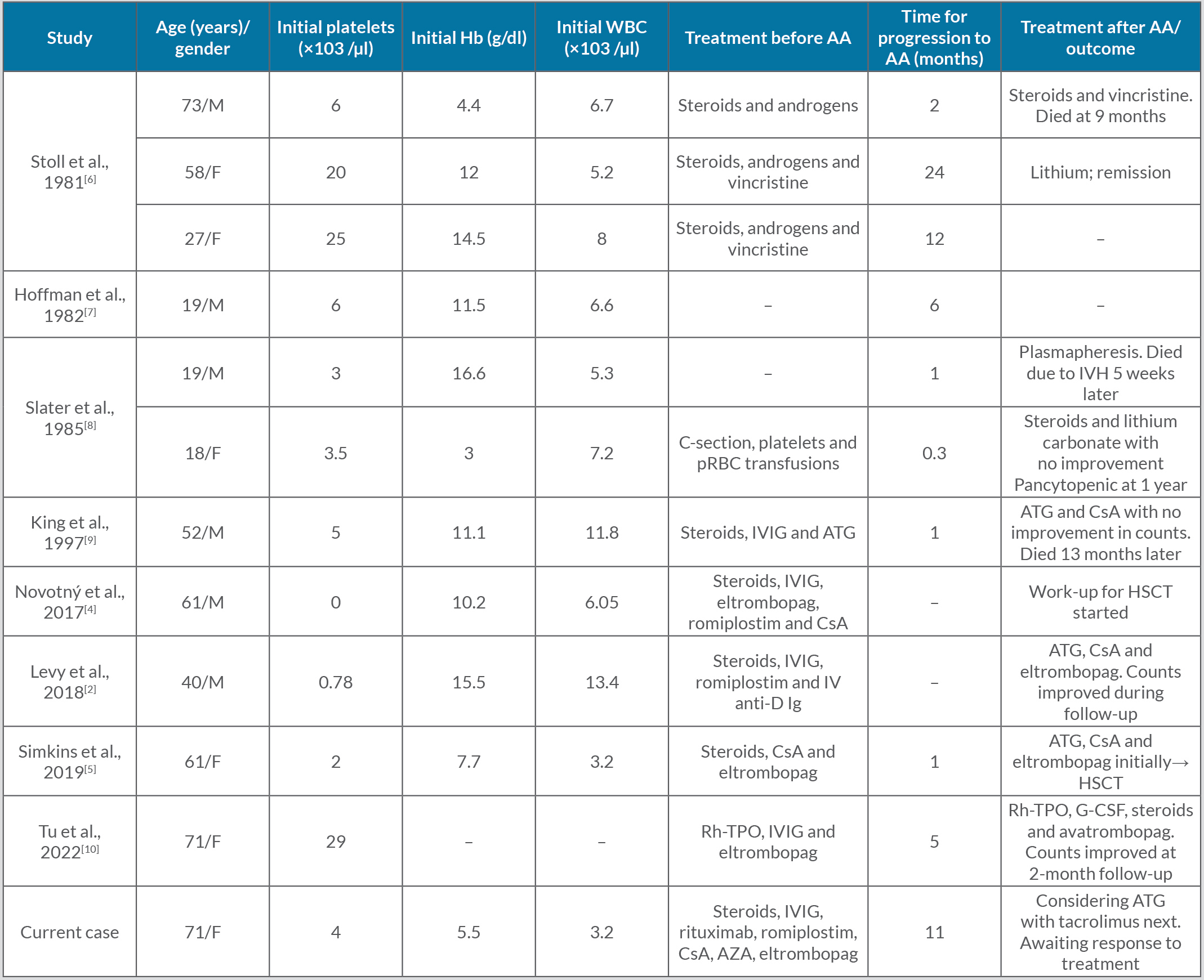
Table 1 (click to enlarge)
Table 1. All cases of reported acquired amegakaryocytic thrombocytopenia progressing to aplastic anaemia, from 1981 to 2022, with therapeutic options.
AA, aplastic anaemia; anti-D Ig, Rho (D) immunoglobulin; ATG, anti-thymocyte globulin; AZA, azathioprine; CsA, cyclosporine A; G-CSF, granulocyte-colony stimulating factor; Hb, haemoglobin; HSCT, haematopoietic stem cell transplant; IVIG, intravenous immunoglobulin; IVH, intraventricular haemorrhage; pRBC, packed red blood cells; Rh-TPO, recombinant human thrombopoietin; WBC, white blood cell count.