ABSTRACT
A 31-year-old Caucasian woman of South-American descent was diagnosed with a variant of multicentric Castleman disease (MCD) that has been reported in Japan as Castleman-Kojima disease. This is a systemic inflammatory disorder known as TAFRO Syndrome which includes thrombocytopenia, polyserositis (ascites/pleural effusion), microcytic anemia, myelofibrosis, fever, renal dysfunction and organomegaly, with immunologic disorder, polyclonal hypergammaglobulinemia, and elevated levels of interleukin-6 (IL-6) and the vascular endothelial growth factor present in serum and/or effusions. Optimal therapies are not well established. The patient was treated with methylprednisolone and rituximab. Following the start of treatment, the patient has been asymptomatic for over 8 months. This is one of only a few reports of TAFRO Syndrome in a non-Japanese patient.
LEARNING POINTS
- Castleman Disease (CD) is a lymphoproliferative disorder which may exhibit a progressive, severe and life-threatening clinical course.
- TAFRO Syndrome (thrombocytopenia, anemia, systemic inflammation (ascites/pleural effusion), myelofibrosis, renal dysfunction and organomegaly) is a rare variant of CD first reported in Japan.
- The cause of the disease is unknown. Elevated levels of interleukin-6 (IL-6) and vascular endothelial growth factor in serum and/or effusions are believed to be linked to the pathogenesis.
- In this case, high doses of steroids and rituximab achieved a remission.
- This rare entity is not always confined to those of Japanese descent and can occur in Caucasian patients.
KEYWORDS
Castleman disease, TAFRO syndrome.
INTRODUCTION
Castleman Disease (CD) is an uncommon lymphoproliferative disorder diagnosed by clinical, histopathological and laboratory criteria. It
was first described in 1954 by Benjamin Castleman[1,2].
Two primary clinical presentations were characterized: the unicentric CD (UCD) confined to a single lymph node, and the multi-centric
CD (MCD) which compromises multiple sites[3]. Several studies have indicated that CD is composed of several disease entities, including
idiopathic CD and secondary CD due to human immunodeficiency type-1 (HIV) infection; autoimmune disease-associated lymphadenopathy;
polyneuropathy; anasarca; organomegaly; endocrinopathy; M-proteins and skin lesions (POEMS Syndrome); and non-Hodgkin's
lymphomas[4,5]. Moreover, the involvement of human herpesvirus 8 (HHV-8) infection has been demonstrated in at least 40-50% of MCD unrelated to HIV in western countries[6,7].
Three histopathologic subtypes have been described: hyaline-vascular (HV); plasma cell; and mixed variant. Of the localized forms of CD, the HV type is found in about 85% of cases and the plasma-cell type in 15%[8,9,10].
The HV type is often asymptomatic, whereas the plasma cell type tends to exhibit a more aggressive clinical course with constitutional
symptoms and laboratory abnormalities.
A variant of MCD has been reported in Japan and labeled Castleman-Kojima disease. This is a systemic inflammatory disorder known as
TAFRO Syndrome which includes thrombocytopenia, polyserositis (ascites/pleural effusion), microcytic anemia, myelofibrosis, fever, renal
dysfunction and organomegaly, with immunologic disorder, polyclonal hypergammaglobulinemia, elevated levels of interleukin-6 (IL-6) and
with the vascular endothelial growth factor present in serum and/or effusions[11,12].
We describe the case of a Caucasian woman with Castleman-Kojima disease (TAFRO Syndrome), who was treated with corticosteroid and
rituximab[13].
CASE REPORT
A 31-year-old woman with no significant medical history presented with recurrent episodes of fever, night sweats and dyspnea for 2 months.
Physical examination revealed fever (38-38.5ºC); skin pallor; enlargement of bilateral, lateral and posterior cervical and axillary lymph nodes; anasarca generalized edema; abdominal distention; tense ascites; and hepatosplenomegaly.
The patient’s hemoglobin (Hb) was 6.3 gr/dl, with a low mean corpuscular volume of 76.8fl. White blood cells were 6.9 x 109 /L and platelets
were 69 x 109 /L. Both creatinine (1.9 mg/dl) and urea (150 mg/dl) were elevated. Although the patient’s liver function test was normal,
albumin, serum iron and ferritin were elevated, suggesting a chronic inflammatory illness.
The patient’s C- reactive protein was 17 mg/dl, the erythroid sedimentation rate was 75 mm/h and the direct Coombs' test resulted strongly
positive with normal haptoglobin, panagglutinin immunoglobulin G (Ig-G) antibodies and without evidence of hemolysis.
A Mantoux test was negative as were serum hepatitis B surface antigen (HBsAg), anti- hepatitis C virus (HVC), HHV-8, anti-HIV, Epstein-
Barr virus Immunoglobulin M (EBV IgM), Huddleson test, Chagas hemaglutination and antibody, venereal disease research laboratory
(VDRL) quantification and toxoplasmosis antibody, cytomegalovirus (CMV) anti-IgG and CMV-polymerase chain reaction (PCR).
Anti-DNA antibody, antineutrophil cytoplasmic antibodies (ANCA), rheumatoid factor and serum complement were all normal or negative;
the antinuclear antibody positive speckled pattern was 1/160; the serum total complement was normal; and beta-2 microglobulin (B2M) was
2.4 mg/L. The serum IL-6 was recorded at 60 pg/ml (<4 pg/ml). Urinalysis showed a small amount of proteinuria. An abdominal ultrasound
showed hepatomegaly on the left hepatic lobe at 135 mm and on the right hepatic lobe at 178 mm, with splenomegaly at 133 x 52 mm.
Echocardiography showed normal left ventricular function with 5 mm of pericardial effusion, and a computed tomographic scan (Figs. 1 and
2) revealed pleural effusion, hepatosplenomegaly, ascites, adrenal glands enlarged with increased blood flow velocity in vessels without
retroperitoneal lymph nodes. The ascitic and pleural fluids were both exudates.
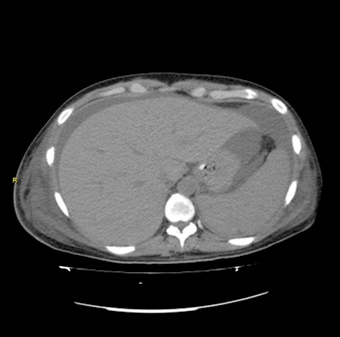
Figure 1 (click to enlarge)
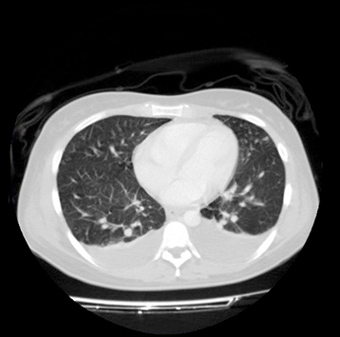
Figure 2 (click to enlarge)
Figure 1: Contrast enhanced computed tomography scan of thorax shows
pleural effusion without lymph nodes.
Figure 2: Contrast enhanced computed tomography scan of abdomen shows
hepatosplenomegaly, ascites, adrenal glands enlarged and flow of blood vessels
increased without retroperitoneal lymph nodes
A right axillary lymph node biopsy is shown in Fig. 3. The immunohistochemical inspection resulted negative for CD20, PAX5 y CD78a, Bcl-2, CD30, CD15 y Alk in follicular region and lymphocytes-T positive for CD3, CD5, CD43, CD30+ and CD79a in the interfollicular area. A bone marrow biopsy showed normal morphology and adequate marrow iron, and the reticulin stain showed grade I fibrosis. The HHV-8 marker was negative.
The patient was transfused with units of packed erythrocytes, and treated with 500 mg/d IV of methylprednisolone and 375 mg/m2 of rituximab, once weekly for 4 doses.
The patient has remained asymptomatic for over 8 months.
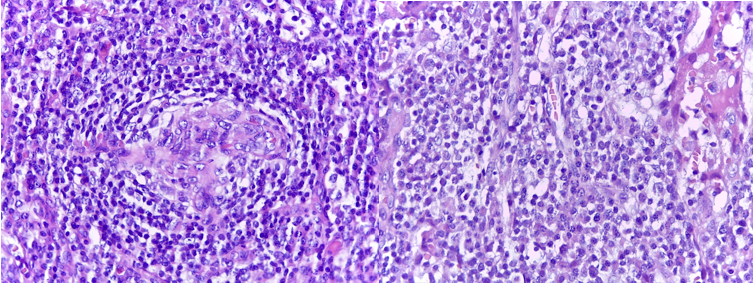
Figure 3 (click to enlarge)
Figure 3: Lymphoid proliferation where the follicles were regressed with interfollicular region and paracortical area, expanded with lymphocytes distributed as “onionskin” fashion with follicular dendritic cells, plasma cells and variable blood vessels. H&E 250x
DISCUSSION
The Castleman-Kojima disease occurs in the middle aged, without any prior history of autoimmune diseases and is four 4 times more common in women than in men.
Kojima et al. reported that MCD in Japan is a hyper-IL-6 syndrome and usually occurs without either HIV or HHV8 infection. It exhibits a chronic disease course, and the authors published 7 cases in which effusions were the initial clinical presentation[14].
The following criteria have been established for the diagnosis of TAFRO syndrome: thrombocytopenia, microcytic anemia, systemic inflammation (Ascites/pleural effusion), myelofibrosis, fever, renal dysfunction and organomegaly. Antinuclear antibody and immunologic disorder (rheumatoid factor, platelet-associated IgG, anti-thyroid antibody) may also appear during the course of the illness[15].
The treatment of CD is not well established. Steroids are commonly used and can achieve remission in up to 60% of cases, however recurrence rates are high. The role of IL-6 in the pathogenesis of the disease has prompted the use of the anti-receptor antibody IL-6 tocilizumab, which has been reported to induce complete remission in several cases[16,17].
The monoclonal anti-CD20 antibody rituximab has also been successfully used, either alone or in combination with chemotherapy[18]. In the present case, rituximab associated with steroids resulted in complete remission. Treatment with chemotherapy (e.g. cyclophosphamide, vincristine, doxorubicin and prednisone or dexamethasone)
also has been reported to achieve high rates of remission, however it provides little chance of recurrence-free survival. Durable responses occur in only 25% of cases, but rarely remissions have been sustained for 15 years[16]. Recurrences after remission worsen the prognosis and limit the chances of survival. Further studies to determine the optimal treatment of this condition are needed.
CONCLUSIONS
Our case meets all the criteria for TAFRO Syndrome in a Caucasian patient of South-American descent. Several questions remain regarding the mechanism and etiology of this rare disease. It is still controversial whether TAFRO Syndrome should be considered as a variant of MCD or if it is a distinct entity. Clinical presentation is dominated by serositis associated with thrombocytopenia, it is not associated with HHV-8 infection, and it has a chronic indolent clinical course.