ABSTRACT
Objectives: To highlight systemic involvement features in Sjögren Syndrome (SS).
Materials and methods: A case of a 32-year-old woman presenting with flaccid tetraparesis, in the setting of severe hypokalaemia, is described.
Results: Additional evaluation confirmed primary SS with type 1 renal tubular acidosis (RTA1) and gammaglobulin-mediated vasculitis. A significant clinical improvement was achieved following adequate treatment.
Conclusion: Extra-glandular involvement in SS is usually due to autoimmune lymphocytic infiltration and severe complications can be avoided if there is a prompt diagnosis.
LEARNING POINTS
- Sjögren Syndrome (SS) can lead to cutaneous involvement (hypergammaglobulinemic vasculitis) and also kidney SS-involvement characterized by renal tubular acidosis.
- The renal tubular acidosis in the presence of SS is usually due to auto-immune lymphocytic tubular infiltration.
- Severe hypokalaemia, nephrolithiasis, nephrocalcinosis, and chronic renal failure are among SS-associated type 1 renal tubular acidosis (RTA1) complications. These can be avoided if there is a prompt diagnosis leading to potassium supplementation, alkali replacement therapy and immunomodulation.
KEYWORDS
Sjögren Syndrome, renal tubular acidosis, severe hypokalaemia, nephrocalcinosis
INTRODUCTION
A renal tubular acidosis (RTA) may be suspected in the presence of metabolic acidosis and hypokalaemia, secondary to high urinary excretion of potassium as a natural renal buffering mechanism. In particular, the distal form (type 1) is associated with diminished H(+)-ATPase activity, impairing the ability to maximally acidify the urine. It usually improves with alkali supplementation, contrary to the proximal RTA (type 2).
Approximately 10% of patients with primary Sjögren Syndrome (SS) have clinically significant kidney involvement. Of these, 73% have type 1 RTA (RTA1), and less frequently, type 2 RTA (RTA2)[1]. In the former, the activity of H(+)-ATPase pump in the collecting duct is totally absent by unknown immunological mechanisms, and the metabolic acidosis may be aggravated by autoantibodies directed against the carbonic anhydrase II[2]. The Authors report a case of hypokalemic paralysis, leading to the disclosure of previously unrecognised RTA1 and immune-mediated vasculitis with accompanying sicca symptoms, as part of SS.
CASE REPORT
A 32-year-old Caucasian woman presented to the emergency department with rapidly onset tetraparesis and palpitations. She had an unremarkable personal history, except for chronic iron anaemia due to menstrual losses. She was not taking any long-term medication. A positive history of chronic kidney disease was present in her father, two paternal uncles, one paternal aunt, and one paternal cousin. On physical examination she had low body mass index (15), showed signs of dehydration, anaemic conjunctivae, slightly swollen parotid glands and lower limb petechiae (Fig. 1).
Her neurological exam revealed flaccid lower limb grade 1 and upper limb grade 3 strength and osteotendinous hyperreflexia, without any meningeal or focal signs. Laboratory tests revealed severe hypokalaemia (1.4mmol/L), hyperchloremic metabolic acidemia with compensatory hyperventilation (arterial pH 7.18; bicarbonate 10 mEq/L; Cl- 128 mEq/L, PCO2 21 mmHg, anion gap 4 mmol/L), and normocytic normochromic anaemia (Hb 9.5 g/dL, MGV 81 fL), in the presence of dilute alkaline urine (density 1006, pH 8.0) and normal kidney urea and creatinine levels. The electrocardiogram (ECG) showed frequent ventricular ectopic beats with right bundle block pattern, bigeminism and prominent U waves (Fig. 2a).
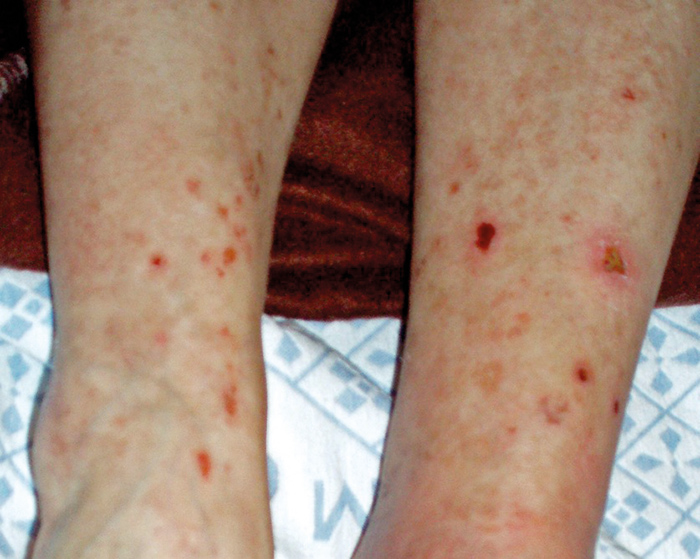
Figure 1 (click to enlarge)
Figure 1: Inferior limb examination revealed scattered single and confluent petechiae in different reabsorption stages.
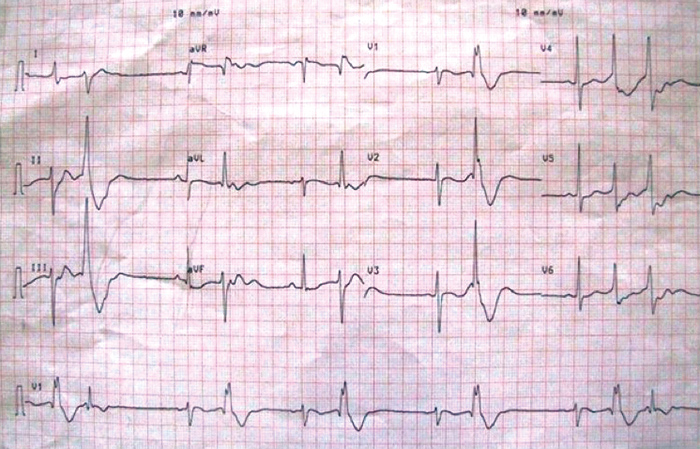
Figure 2A (click to enlarge)
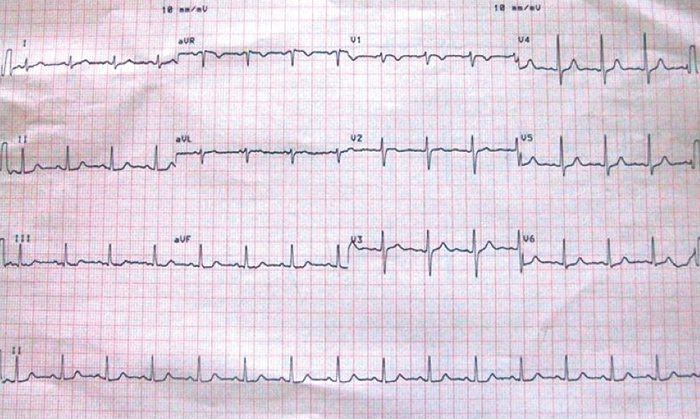
Figure 2B (click to enlarge)
Figure 2A: ECG recording showing sinus rhythm with bigeminism, plus frequent ventricular ectopic beats with right bundle block pattern and prominent U waves.
Figure 2B: ECG recording showing normal sinus rhythm after potassium correction.
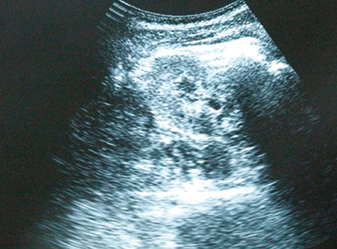
Figure 3A (click to enlarge)
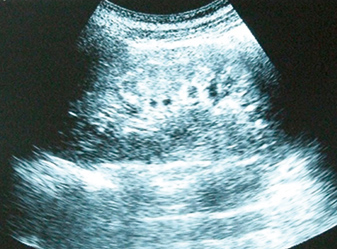
Figure 3B (click to enlarge)
Figure 3A-B: Renal ultrasonogram showing normal-sized kidneys with hyperechogenic areas in the medulla of both kidneys, indicating medullary nephrocalcinosis.
After intravenous fluid challenge and potassium correction, she recovered muscular strength and the ECG results normalized (Fig. 2b).
On subsequent hospital days, however, sustained hypokalemia (< 3.5 mEq/L), with inappropriately high urinary transtubular potassium gradient (7.1) despite intravenous potassium supplementation, metabolic acidosis and alkaline urine pH (> 7.3) with positive urine anion gap (19 mEq/L), suggested RTA1. The tubulopathy was confirmed by impaired urinary acidification following an ammonium chloride load test (Table. 1).
The tubular defect was accompanied by mild hypocalcaemia (total calcium of 8.5 mg/dL; ionized calcium of 2.1 mmoL/L; normal, 2.26-2.64 mM/L) and nephrocalcinosis (Fig. 3), but remarkably without associated hypercalciuria (4.8 mmoL/24h, normal 2.5-8) or increased PTH (27.8 pg/mL, normal 14-72). A glomerular filtration rate of 80 mL/min with mild proteinuria (0.36 g/24h, normal <0.15) revealed an already established KDOQI stage 2 chronic renal failure.
| Time after loading (h) | 2 | 4 | 6 |
| Volume (urine) (mL) | 40 | 40 | 240 |
| pH (urine) | 8.07 | 7.81 | 7.65 |
| Amonium (urine) (mmol/L) | 125 | 125.7 | 128.9 |
| pH (blood) | 7.328 | 7.258 | 7.311 |
Table 1: Ammonium chloride acidification test showed impaired urinary acidification.
On further questioning, the patient had admitted to experiencing xerophthalmia, xerostomia with polydipsia, relapsing parotid gland swelling, lower limb purpura and dyspareunia for 12 years. She never sought medical attention since these complaints never impaired her daily life activities, except for the need to drink 3-4L of water per day to quench her thirst, with subsequent polyuria. The documented increased natriuresis (416 mmoL/24h, normal 40-220), kaliuresis (114.8 mmol/24h, normal 25-125) and urinary osmolality (1194 mOsmol/Kg, normal 300-900), were in concordance with these symptoms. Asthenia, anorexia, weight loss (2 kg) and incomplete Raynaud phenomenon had begun 3 months prior. The clinical presentation suggested an impaired exocrine gland secretion. The sicca diagnostic findings included positive antibodies to the ribonucleoprotein antigen La (SS-B) and Ro (SS-A), antinuclear antibody (ANA) 1:320 with granulated pattern, and negative results in rheumatoid arthritis (RA) test, anti-DNAds, anti-Sm, anti-SCL70 and anti-centromerase antibodies. Severe hypofunction of both submandibular glands was documented (Fig. 4).
The parotid gland ultrasonography documented symmetric hypoechoic glands with normal dimensions (Fig. 5) and its biopsy reported the presence of diffuse lymphoplasmocytic infiltration. Serum beta 2 microglobulin was within the normal range.
Dry eye was confirmed by Schirmer’s test (RE 12 mm/5min, LE 15 mm/5 min), but there was no sign of keratoconjunctivitis (Rose-Bengal test). Vasculitis was likely secondary to hypergammaglobulinemia (18.6g/L with immunofixation in the serum of IgG 19.90 g/L and l 2.85 g/L). The anaemia workup revealed decreased serum iron (14 ug/dL) and ferritin (35.3 ng/mL) levels, with no suggestive features of celiac disease present. Haemolysis parameters, such as lactate dehydrogenase (LDH), haptoglobin, bilirubin and Coombs test, were unremarkable.
Therefore, hypergammaglobulinemic purpura and secondary RTA1 due to SS with concomitant iron anaemia were diagnosed. Subsequently, corticotherapy and azathioprine was started. Oral intake of potassium citrate and potassium chloride for correction of hypokalaemia, and adequate alkali supplementation with sodium bicarbonate were also initiated. Oral iron supplementation, artificial and oral hydration ad libidum were also prescribed. Since then, hypokalemia and metabolic acidosis were corrected, and the patient mentioned symptomatic relief and body mass index (BMI) increase.
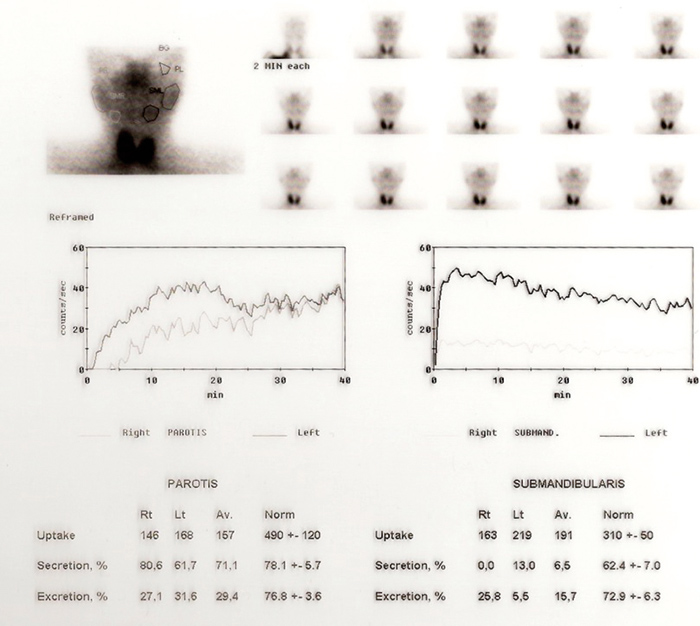
Figure 4 (click to enlarge)
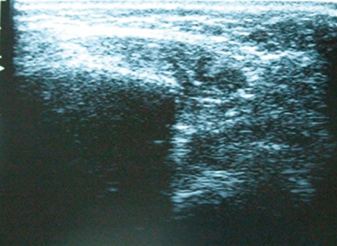
Figure 5 (click to enlarge)
Figure 4: Parotid gland ultrasound showed symmetric normal-sized hypoechoic glands.
Figure 5: Salivary scintigram showing almost no uptake and reduced excretion in the salivary glands (class IV Schall).
DISCUSSION
This patient met all primary SS diagnostic criteria of the American-European Consensus Group (AECG) classification[3], published in 2002, and of the more recent Sjogren's International Collaborative Clinical Alliance classification criteria[4]. This disease is characterised by exocrine glandular autoimmune lymphocytic infiltration, with varying degrees of systemic involvement. Our patient presented cutaneous (hypergammaglobulinemic vasculitis) and kidney involvement. The latter presented as an interstitial nephritis, disclosed by distal RTA. In an attempt to buffer blood pH, calcium salts are continually mobilized from bone, while metabolic acidosis suppresses proximal and distal calcium reabsorption by decreasing apical calcium entry, leading to hypocalcaemia, hypercalciuria and low urinary citrate excretion. These patients are prone to develop nephrocalcinosis, osteomalacia and compensating secondary hyperparathyroidism. Nevertheless, as in the present report, nephrocalcinosis can occur in RTA1 patients without hypercalciuria contribution[5], with hypocitraturia and persistent alkaline urine concurrently accounting for the calculi development. Potassium citrate administration corrects the metabolic acidosis, resulting in increased citrate excretion levels, making it the best alkali supplement, correcting both hypokalaemia and hypocitraturia.
Potassium bicarbonate may be equally effective for the therapy of RTA1, however, it is associated with side effects such as gas formation of CO2 in the stomach. Nonetheless, treatment should be aimed at the immunological disease and work to decrease the lymphocytic infiltration of the exocrine glands and extra-glandular tissues, at least, partially to correct the acidification defect, improve bone metabolism and improve overall length and quality of life of primary SS patients.