ABSTRACT
Phenylketonuria (PKU), the most common inborn error of amino acid metabolism, has been associated with an increased risk of renal impairment, the reason for which is not fully understood. We report the case of a 33-year-old female patient diagnosed with PKU in adulthood after suspicion of the disease in her daughter. The diagnostic approach revealed proteinuria, and minimal change disease (MCD) was identified. Therapeutic measures and follow-up are discussed. The possible link between these two disorders is interesting. Attenuated developmental delay of untreated PKU could be explained by the decreased accumulation of phenylalanine secondary to proteinuria. On the other hand, MCD may be a possible, although as yet unreported, pathophysiological mechanism explaining the renal repercussions that have been found in adult PKU patients, who should be screened for proteinuria.
LEARNING POINTS
- Phenylketonuria (PKU) is associated with renal impairment so patients with PKU should be periodically screened for proteinuria because podocytopathies, such as minimal change disease (MCD), may be an underlying misdiagnosed condition.
- MCD in adults demands tight surveillance in case of severe complications including nephrotic syndrome, haematuria and acute kidney injury, and although remission with glucocorticoid therapy is typical, 50–75% of patients will relapse.
- MCD may be a previously unreported cause of proteinuria and renal involvement in adult patients with PKU, where control of hypertension and moderation of protein overload are essential.
KEYWORDS
Minimal change disease (MCD), proteinuria, nephrotic syndrome, phenylketonuria (PKU), phenylalanine
CASE DESCRIPTION
We describe a 33-year-old female patient evaluated by an internal medicine consultant due to the possibility of hereditary and/or metabolic disease after global developmental delay was detected in her 6-month-old daughter. Maternal PKU was suspected because of the association between global psychomotor developmental delay, microcephaly and a family history of cognitive impairment. A serum aminogram uncovered a remarkably elevated phenylalanine (Phe) level of 1,663 nmol/ml (normal range 50–90 nmol/ml) with reduced tyrosine, confirming the diagnosis of classic PKU. The patient’s medical history included non-medicated arterial hypertension and learning difficulties in school.
On physical examination, the patient presented with peripheral oedema and a BMI of 28 kg/m2, without any other significant findings. Neuropsychological evaluation revealed alterations in executive functions as well as visual-perception and constructive impairment. Brain T2-weighted magnetic resonance imaging (MRI) showed widespread high-intensity areas in the deep white matter, especially the bilateral occipital lobes (Fig. 1). Laboratory findings including a complete blood count, renal and hepatic function were unremarkable.

Figure 1 (click to enlarge)
Figure 1. Axial T2-weighted MR images showing the symmetrical increase in the T2-weighted signal in the periventricular white matter, especially in bilateral occipital lobes, common in phenylketonuria
The patient started antihypertensive medication and dietary restriction of Phe, but maintained poor therapeutic compliance, with Phe concentrations above the recommended level of 120–600 µmol/l. She also maintained uncontrolled hypertension, requiring three antihypertensive drugs (including an ACE inhibitor), while 24-hour urine documented proteinuria of 1.4–1.6 g/day, with normal levels of urea (10 mg/dl; normal 6–23 mg/dl), creatinine (1.01 mg/dl; normal 0.50–1.20 mg/dl) and albumin (3.7 g/dl; normal 3.5–5.2 g/dl). A complete infectious and autoimmune study was requested because of the proteinuria.
Further serological studies including a viral hepatitis panel, HIV, anti-nuclear antibody, rheumatoid factor, anti-Ds-DNA, anti-ANCA and anti-citrullinated peptide antibody and urinary eosinophils were negative. C3/C4 were normal. Serum protein electrophoresis revealed hypogammaglobulinemia. Phospholipase A2 receptor antibodies were negative. Renal ultrasound did not show any relevant changes.
Therefore, in light of the still unexplained proteinuria, a renal biopsy was performed which exhibited 20 glomeruli without mesangial expansion or increased cellularity by light microscopy and the absence of complement or immunoglobulin deposits by immunofluorescence microscopy (Figs. 2 and 3). Electron microscopy revealed diffuse effacement of the podocyte foot processes, supporting the diagnosis of minimal change disease (MCD) (Fig. 4).
The treatment approach included optimising hypertension control, reinforcing the dietary restriction of Phe, and stopping consumption of synthetic amino acid (AA) formulas. In light of the normal renal function and mild level of proteinuria, our patient did not immediately start glucocorticoid therapy but was kept under tight surveillance for 18 months. Although further studies revealed proteinuria of less than 1 g/day, a glucocorticoid regimen is now being planned.
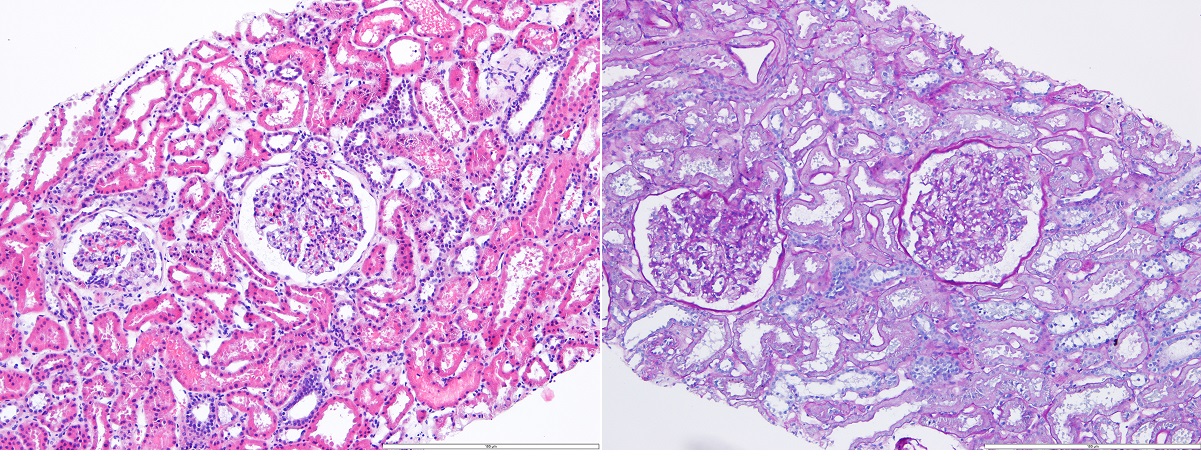
Figure 2 (click to enlarge)

Figure 3 (click to enlarge)
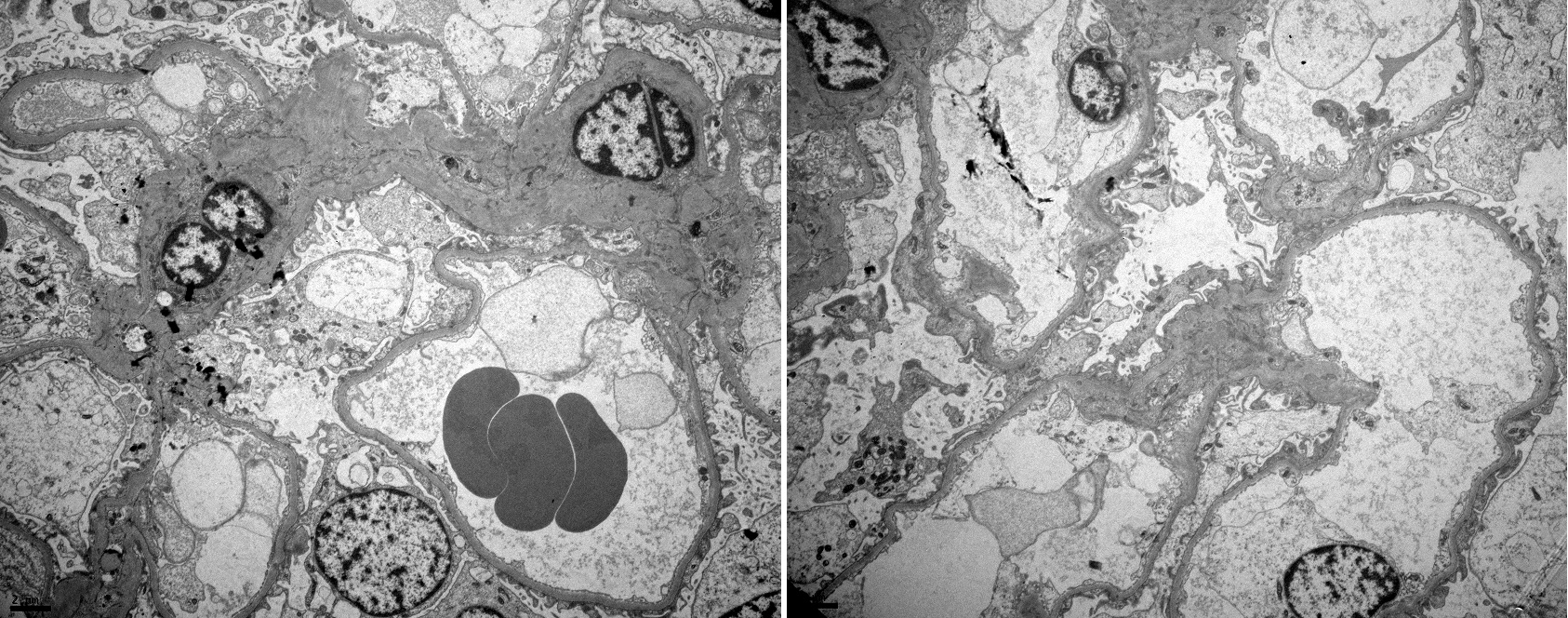
Figure 4 (click to enlarge)
Figure 2. Renal biopsy revealing renal cortex with slight lesions indicating acute tubular damage and without other significant changes, including the absence of complement or immunoglobulin deposits by immunofluorescence microscopy (IgA, IgG, IgM, C3c, C1q and fibrinogen) (left: HE 100×, right: PAS 100×)
Figure 3. Light microscopy showing glomeruli with open capillary loops and no evidence of cellular crescents, fibrinoid necrosis or endocapillary hypercellularity
Figure 4. Electron microscopy disclosing diffuse effacement of the podocyte foot processes, the typical histological lesion of minimal change disease
DISCUSSION
Phenylalanine hydroxylase (PAH) deficiency, otherwise known as phenylketonuria (PKU), is the most frequent inborn error of amino acid metabolism. It is an autosomal recessive disorder which if left untreated results in global developmental delay and severe intellectual impairment [1].
We report the unexpected diagnosis of MCD in an adult patient with PKU, after proteinuria was investigation in a complex diagnostic approach. The association of these two rare disorders is unique and although it may be a coincidence, patients with PKU are known to have an increased risk of renal impairment, the reason for which is not fully understood [2, 3]. Considering the difficulty in studying longitudinal outcomes in rare diseases, the causes of renal disease cannot always be thoroughly investigated. Moreover, renal disease in these patients could easily be missed, especially in the early stages.
We highlight the fact that although proteinuria is a common condition associated with several disorders, in our case a specific disease was diagnosed with significant implications for treatment and follow-up. Furthermore, it is easy to test for proteinuria and these patients should be periodically screened because podocytopathies like MCD may be an associated misdiagnosed condition.
A few published reports have described somatic comorbidities in PKU patients, such as renal impairment, proteinuria, arterial hypertension and obesity [2, 3]. According to the literature, PKU is a potential cause of tubulointerstitial disease associated with increased oxidative stress, local damage due to high Phe excretion and the highly concentrated AA used as a complement in Phe-restricted diets, which lead to greater renal workload, thereby affecting kidney function. Indeed, after MCD diagnosis, the suspension of synthetic AA consumption resulted in reduced proteinuria in our patient [2, 3].
This case is remarkable both because of the rarity of a delayed PKU diagnosis after the implementation of the newborn blood spot screening programme (Fig. 5), and also because of the few signs of intellectual impairment presented by this adult patient despite identification of the classic form of PKU. The attenuated developmental delay due to untreated PKU in this patient could be explained by the decreased accumulation of Phe secondary to proteinuria associated with MCD. Plasma and urine amino/organic acid analyses should be carried out in adult patients with learning difficulties and unexplained neuropsychological manifestations.

Figure 5 (click to enlarge)
Figure 5. Newborn blood spot screening card, including phenylketonuria (since 1966) congenital hypothyroidism (since 1977), cystic fibrosis (since 1988) and various inborn errors of metabolism (since 2002)
MCD may be secondary to drug intake, neoplasms, infections and allergies, or else primary, which is probably the case in this patient[4]. The pathogenesis underlying MCD is unclear. MCD is a major cause of nephrotic syndrome (approximately 90%) in children and in a minority of adults (approximately 10%) [4]. We underline the early identification of MCD in this patient, explaining the mild proteinuria levels documented so far. Moreover, we highlight the importance of surveillance for serious manifestations of MCD, such as nephrotic syndrome, haematuria and acute kidney injury [4, 5].
We emphasise the importance of blood pressure control and stopping consumption of synthetic AA formulas. The mainstay of therapy for MCD is prednisolone, with >70% of adults, particularly younger patients, achieving complete remission within 18 months [4, 5]. Although 5–10% of untreated patients can spontaneously remit within 3 months, according to the literature, when persistent nephrotic syndrome is evident, glucocorticoid therapy should not be withheld due to possible thrombotic and infectious complications [4, 5].
PKU presents with unclear multisystemic manifestations which are currently under study, especially in adult patients. Investigation of the link between PKU and MCD could provide better understanding of the pathophysiology of both disorders as protein homeostasis is affected in each condition. Moreover, there are therapeutic and follow-up implications for both disorders and affected patients should be screened for proteinuria.