ABSTRACT
Lisinopril is an angiotensin converting enzyme inhibitor (ACE-I) that has been on market for more than 25 years. ACE-I are usually well tolerated and rarely have serious or life-threatening side effects. We describe an unusual presentation of fulminant hepatic cholestasis probably secondary to lisinopril. To our knowledge, this is the second case report which shows lisinopril-induced liver injury though a cholestatic mechanism. The patient was a 59-year-old woman with type 2 diabetes, a high body mass index and hypertension, who presented with a 5-week history of jaundice and itching. She had been started on lisinopril for diabetic nephropathy 8 weeks before admission. Other causes for cholestasis had been excluded through non-invasive immunology and virology screening, an ultrasound of the liver, magnetic resonance cholangiopancreatography and a liver biopsy. The biopsy was consistent with drug-induced liver injury. Lisinopril was stopped 2 weeks before admission. The patient’s hospital stay was complicated by contrast nephropathy and influenza A which were both treated appropriately. Unfortunately, the liver cholestasis did not completely resolve following withdrawal of lisinopril and the patient died after 4 months. A literature search yielded only six other reported cases of lisinopril-induced liver injury. Five cases described hepatocellular damage and one showed cholestatic injury.
LEARNING POINTS
- Angiotensin converting enzyme inhibitors (ACE-I) rarely have serious or life-threatening side effects.
- Lisinopril-induced liver injury can present as hepatocellular or cholestatic injury.
- Severe hepatotoxicity secondary to lisinopril can be life threatening irrespective of the liver injury pattern.
KEYWORDS
Angiotensin converting enzyme inhibitors, lisinopril, drug-induced liver injury, cholestasis
CASE DESCRIPTION
A 59-year-old woman presented with a 5-week history of obstructive jaundice (dark urine, pale stools and itching). She had type 2 diabetes mellitus with peripheral neuropathy and nephropathy, hypertension, polymyalgia and chronic fatigue. She had no previous or family history of liver problems.
She had started lisinopril for diabetic nephropathy (creatinine 130 µmol/l (N 44–80 µmol/l) with albuminuria) 8 weeks previously. After 3 weeks, she reported icteric eyes and skin to her general practitioner who stopped the lisinopril. She denied excessive alcohol intake and over-the-counter drug use. Her long-term repeat medications were betahistine, metformin, paroxetine, prochlorperazine, atorvastatin and gliclazide. No other prescribed medication had been started during the previous year.
On examination, the patient was alert, had a high body mass index (108 kg) and was jaundiced, without other stigmata of liver disease, and the abdomen was normal. Tests showed initial serum bilirubin 93 µmol/l (N <21 µmol/l), alanine transaminase (ALT) 92 IU/l (N <33 IU/l), alkaline phosphatase (ALP) 1,916 IU/l (N 30–130 IU/l), prothrombin time (PT) 30 seconds (N 9.4–11.4 seconds), corrected from 30 to 12.8 seconds after vitamin K administration, white cell count 10.5×109/l (N 3.5–9.5×109/l), eosinophils 0.22×109/l (N 0.04–0.5×109/l) and creatinine 105 µmol/l. Her liver function tests (LFT), before the initiation of the lisinopril, were within the normal ranges. Serum was negative for hepatitis A, B, C, E, EBV, CMV and HIV markers and also for antinuclear, smooth muscle, and liver kidney microsomal and mitochondrial antibodies. Serum IgA was mildly elevated at 4.9 g/l (N 0.8–4.0 g/l), while serum IgG, IgM, alpha-1-antitrypsin, ferritin and serum iron were all within normal ranges. Ultrasound of the abdomen showed fatty infiltration of the liver with a normal biliary tree. Magnetic resonance cholangiopancreatography confirmed no intra- or extrahepatic biliary dilatation.
A liver biopsy was performed 7 days after admission (Figs. 1 and 2). The peri-portal areas showed cholestasis with canalicular bile plugs, which was acute as no copper or copper-binding protein was seen (Fig. 2), and fibrosis, with focal portal-to-portal bridging fibrosis (Fig. 1) was also present. There was also minimal centrilobular steatosis. The findings were consistent with drug-induced liver injury (DILI). Fatty liver disease was not demonstrated on this liver biopsy.
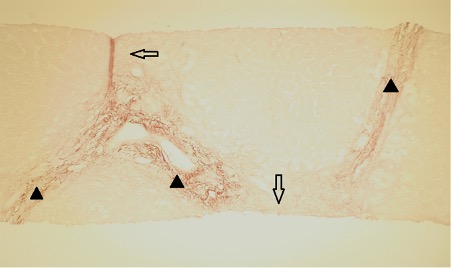
Figure 1 (click to enlarge)
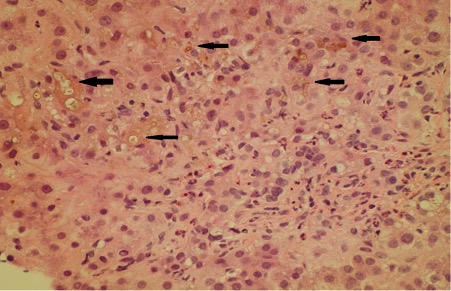
Figure 2 (click to enlarge)
Figure 1. Portal fibrosis and bridging fibrosis (Orcein stain). Black arrowheads indicate original portal areas, while clear arrows indicate new elastic fibres.
Figure 2. Cholestatic hepatitis (H&E stain). Canalicular cholestasis is indicated by arrows (yellow-green pigment)
Contrast computed tomography (CT) was performed to rule out any serious internal pathology as the patient had been referred for consideration for liver transplantation. Despite appropriate pre- and post-CT scan precautions, she developed contrast nephropathy (creatinine rising to 335 µmol/l) and needed dialysis for 4 weeks. She also developed influenza A which was treated with oseltamivir.
Although the lisinopril had been stopped 5 weeks before admission, LFT initially worsened with bilirubin rising to 270 µmol/l, PT was 12.6 seconds, and ALT, AST, ALP and GGT were all elevated. The patient was referred to the regional liver transplant centre, but was deemed unsuitable for transplantation as she was so unwell with a poor baseline functional status.
Subsequently, LFT improved modestly (ALP 963 IU/l). After 3 months in hospital, the patient was discharged to a rehabilitation centre to improve her fitness for liver transplantation. The jaundice persisted (bilirubin hovering between 170 and 200 µmol/l), with ALP and GGT remaining elevated. When seen in the clinic 4 weeks after discharge, the patient had deteriorated and was unable to engage with rehabilitation. She became completely bed bound and died shortly afterwards.
DISCUSSION AND LITERATURE REVIEW
ACE-I are frequently used in patients with hypertension, diabetic nephropathy and after myocardial infarction[1]. Severe side effects associated with ACE-I are uncommon. The reported case showed a rare pattern of ACE-I-induced liver injury.
DILI is usually a diagnosis of exclusion. Improvement in LFT after stopping the drug is typical but not invariable. Biochemically, DILI can be of one of three types: hepatocellular, cholestatic or mixed pattern. The type is determined by the equation: Ratio (R)=ALT (measured in multiples of upper limit of normal)/ALP (measured in multiples of upper limit of normal). DILI is considered hepatocellular if R is more than 5, cholestatic if R is less than 2 and mixed if R is between 2 and 5[2].
Cholestatic liver injury has been reported with several other ACE-I, including enalapril, ramipril and fosinopril[3]. A PubMed and Medline search yielded only six other reported cases of lisinopril-induced liver injury [1, 4–8]. Of these, five showed hepatocellular damage and one showed cholestatic injury[1]. No patient had peripheral eosinophilia which is generally associated with hypersensitivity reactions[8].
Table 1 provides details of the present and previously published cases of lisinopril-induced liver injury.
Liver histology was available in five patients. All, including the present case, developed lymphocytic or monocytic portal or peri-portal hepatitis[1, 4, 7, 8]. Histological cholestasis was present in four cases[1, 4, 7] and three cases showed evidence of portal fibrosis [1, 7]. Centrilobular necrosis was seen in one case only [4]. None of the patients had eosinophilic infiltration.
In three patients, the liver injury resolved. Three other patients (including the one reported here) died and one developed chronic liver disease [1, 4–8].
Our patient had not started any prescribed or over-the-counter medication, other than lisinopril, during the year before admission. Therefore, the derangement in LFT due to cholestasis was most probably caused by lisinopril. It is unlikely that the patient’s other regular medications, especially gliclazide and atorvastatin, would have precipitated cholestasis. First-generation sulfonylureas are associated with cholestatic liver injury, but gliclazide is a second-generation sulfonylurea. Three case reports have described gliclazide-induced acute hepatitis, but all within 3 months of commencing the drug, and patients seemed to improve after discontinuing the medication[9].
Previous studies revealed evidence of atorvastatin hepatotoxicity in the form of mildly raised ALT. Severe hepatotoxicity is a very rare side effect of atorvastatin. Severe hepatotoxicity secondary to atorvastatin usually manifests as mixed cholestatic/hepatocellular injury. The time between drug exposure and presumed atorvastatin-induced liver injury was 3 months or less in a number of cases reported in the English literature[10].
CONCLUSION
We report a second case of lisinopril liver injury, with a cholestatic liver enzyme pattern, which unfortunately did not resolve.