ABSTRACT
Pituitary apoplexy is a rare endocrine emergency, characterized by a sudden increase in pituitary gland volume secondary to acute ischaemic infarction or haemorrhage of the pituitary gland, usually in the presence of a pituitary adenoma. We present the case of a 79-year-old man admitted for new-onset, bi-temporal and severe headache, associated with photophobia and vomiting, whose additional study revealed pituitary apoplexy. This case highlights the need for high clinical suspicion of this rare entity in order to reduce the associated mortality.
LEARNING POINTS
- Pituitary apoplexy can present with severe headache without ophthalmoplegia or impairment of consciousness.
- It may be mistaken for temporal arteritis.
- CT may be normal so MRI is the diagnostic imaging of choice.
KEYWORDS
Pituitary apoplexy, adenoma, corticosteroids
CASE DESCRIPTION
A 79-year-old man was admitted to hospital with new-onset, bi-temporal and severe headache, associated with photophobia and vomiting. The patient denied visual disturbances, jaw claudication and other focal neurological symptoms. He was fully conscious, afebrile and haemodynamically stable. Funduscopic examination showed a well-defined optic disk and a visible bilateral venous pulse. The patient’s erythrocyte sedimentation rate was high (84 mm/h) and serum sodium level was low (133 mmol/l). A brain CT scan was normal.
Given the clinical suspicion of giant cell arteritis, despite the need for exclusion of cerebral venous thrombosis, the patient started prednisolone 40 mg per day and acetylsalicylic acid 100 mg per day, and a temporal artery biopsy was scheduled. MRI showed the pituitary gland was slightly increased in size and pituitary function tests indicated low ACTH, LH, prolactin and testosterone (Table 1). MRI directed to the sella turcica showed the gland was increased in size due to an enlarged adenohypophysis, with alterations compatible with pituitary apoplexy, but the optic chiasm was preserved (Fig. 1). Temporal artery biopsy failed to find any lesions compatible with giant cell arteritis. The patient stopped prednisolone and started hydrocortisone, with careful control of fluids and electrolyte balance and conservative non-surgical management.

Figure 1 (click to enlarge)
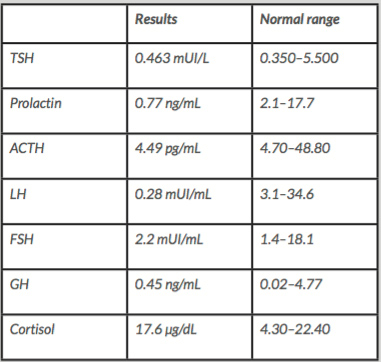
Table 1 (click to enlarge)
Figure 1. Non-enhanced MRI image (T1) showing a large pituitary gland mainly due to an enlarged adenohypophysis
Table 1. Results of baseline anterior pituitary function tests
DISCUSSION
Pituitary apoplexy is a potentially life-threatening condition caused by infarction or haemorrhage of the pituitary gland. Pearce Bailey described the first case of pituitary tumour-associated haemorrhage in 1898, but the term pituitary apoplexy was first coined by Brougham et al. in 1950. Pituitary apoplexy occurs in 0.6–10% of treated pituitary adenomas, and 0.2–0.6 events occur per 100 person-years in non-functioning pituitary adenomas.
Most patients present in the fifth or sixth decade of life and there is a slight male preponderance ranging from 1.1 to 2.25:1.0. Mostly, the primary event involves an adenoma, but pituitary apoplexy may also occur in non-adenomatous or even a normal pituitary gland, especially during pregnancy. The precise pathophysiology is not completely understood, but a proposed hypothesis involves tumour vascular reduction or occlusion due to tumour growth, with an increase in intrasellar pressure.
Precipitating factors can be identified in only about 40% of cases, with arterial hypertension being the most common predisposing factor[1–3].
The clinical presentation is variable, with the most frequent presentation being severe headache, visual defects, ophthalmoplegia and impairment of consciousness. The symptoms are related to anatomical structures in the proximity of the pituitary gland which are prone to compression due to tumour expansion [1,3].
Brain imaging is required to identify a pituitary lesion and although a cranial CT scan is easier to obtain, it is less sensitive for the diagnosis of pituitary lesions, so MRI is the diagnostic imaging modality of choice. Additionally, evaluation of pituitary hormones is required and nearly 80% of patients will have deficiency of at least one of the anterior pituitary hormones at presentation. Clinically, the most important endocrine dysfunction is adrenocorticotropic hormone (ACTH) deficiency, reported in up to 70% of cases.
The immediate medical management of patients with pituitary apoplexy includes careful assessment of fluids and electrolyte balance, replacement of corticosteroids, and supportive measures to ensure haemodynamic stability. Neurosurgical decompression should be considered in patients with severe visual impairment, and long-term hormonal replacement therapy is needed in about 80% of patients [4,5].