ABSTRACT
Objectives: To describe the possible pitfalls in correctly interpreting clinical, radiological and biochemical findings in ACTH-dependent Cushing's syndrome.
Methods: We describe a case of a pituitary adenoma visualized at MRI not correlated with an ACTH-dependent Cushing’s syndrome.
Results: Radiological imaging and hormonal testing can be misleading in suspected pituitary ACTH-related Cushing’s syndrome.
Conclusion: Correct interpretation of the initial clinical presentation can help in the proper diagnosis and treatment of ACTH-dependent Cushing’s syndrome.
LEARNING POINTS
- Occult neoplasia should always be excluded in cases of severe ACTH-dependent Cushing's syndrome.
- A positive MRI result can be misleading.Ectopic ACTH-dependent syndrome is generally associated with a peculiar phenotype.
KEYWORDS
Ectopic ACTH-dependent Cushing's syndrome; endocrinology.
INTRODUCTION
ACTH-dependent Cushing's syndrome (CS) can be difficult to diagnose. Pituitary adenomas represent nearly 70% of all cases of hypercortisolism[1] and about 85% of all CS cases. ACTH-secreting adenomas account for 8-10% of all functioning adenomas and are usually micro-adenomas (95%), rarely exceeding 10 mm in diameter. Imaging (MRI/CT) normally guarantees good characterisation of a pituitary lesion. However, radiological findings can be misleading when used in the diagnostic pathway employed for ACTH-dependent syndromes[2]. The differential diagnosis should, in fact, always consider ectopic ACTH syndrome (EAS), which is normally driven by neuroendocrine tumours (pulmonary, pancreatic or thymus carcinoid, pulmonary microcytoma, medullar thyroid carcinoma or pheochromocytoma)[3]. An occult neoplasia, especially in severe cases presenting with marked muscle weakness, hypokalaemia and hyperpigmentation, could be as present in up to 8% of cases. In addition, the prevalence of pituitary adenomas (about 80% of surgically treated incidentalomas) is rather high[4] and likely to increase over time due to easier access to imaging studies and advances in analytical techniques. About half of these pituitary adenomas prove to be non-functioning tumours, whose presentation is normally secondary to mass-related symptoms; however, incidental findings account for about 10% of cases[5] Therefore, in every endocrinological systemic disease, the possibility of the simultaneous presence of a non-functioning adenoma should always be kept in mind, especially when a pituitary lesion is discovered incidentally. The aim of our study is to describe possible pitfalls in the diagnosis of ACTH-dependent CS.
We describe a case of ACTH-dependent CS, and outline our diagnostic protocols for correctly interpreting the radiological findings according to clinical presentation and hormonal testing. Specifically, we describe the simultaneous presence of a non-secreting pituitary adenoma not correlated with the systemic syndrome.
CASE REPORT
A 48-year-old woman who was a heavy smoker, was admitted to the internal medicine department of our institute with progressive muscle weakness and a 5 kg weight loss over 3 months associated with sweating and peripheral paraesthesia and oedema. The patient’s medical history included anxiety-depressive disorder. On physical examination, she displayed a typical Cushingoid appearance (moon face, proximal muscle hypotrophy, hyperpigmentation, hirsutism, buffalo hump and purpura). Her blood pressure and pulse rate were slightly increased. Laboratory tests showed marked hypokalaemia (1.9 mEq/l) associated with metabolic alkalosis, neutrophil leukocytosis and hyperglycaemia. Her hormonal secretory profile showed abnormal values for PTH (74.4 pg/ml, nv 12–72), calcitonin (320 pg/ml, nv <5.5 pg/ml), gastrin (176 pg/ml, nv <108 pg/ml) and chromogranin (200 µg/ml, nv 19–98). The main tumour markers were negative. Her fasting plasma cortisol serum and 24 h urinary free cortisol values were 59.36 µg/dl (nv 6.2–19.4 µg/dl) and >1000 µg/24 h (nv 32–243 µg/24 h), respectively, and did not decrease after 1 mg and 8 mg overnight dexamethasone. The high ACTH level (165.1 pg/ml, nv 7.2–63.3 pg/ml) excluded primary adrenal causes. The patient thus underwent a pituitary region MRI with contrast medium (1.5 Tesla) which showed an intrasellar pituitary micro-adenoma (4 mm) (Figs. 1 and 2), a CT scan of the abdomen and a chest x-ray which excluded significant changes in the adrenal glands or suspect lesions in other parenchyma. The patient’s clinical condition rapidly worsened with sensorium depression and psychotic behaviour. Dynamic tests were performed in order to confirm the suspected diagnosis of a pituitary ACTH-secreting adenoma. Surprisingly, neither corticotropin-releasing hormone (CRH) nor desmopressin administration significantly increased ACTH or cortisol levels, suggesting an ACTH source other than the pituitary. This suggestion was supported by inferior petrosal sinus (IPS) sampling showing no significant centre-periphery ACTH gradient either at baseline or following CRH stimulation. The search for an ACTH source included an OctreoScan test which was negative, while a whole-body FDG-PET scan revealed an enhancing lesion of the inferior right lobe of the lung, confirmed by a chest CT scan (Fig. 3). Fibre-optic bronchoscopy revealed an endobronchial mass with typical lung carcinoid features.
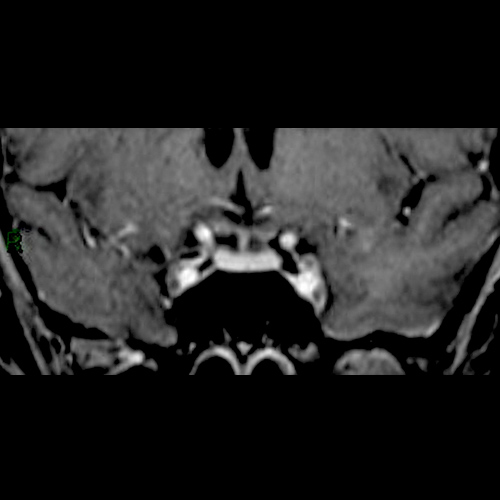
Figure 1 (click to enlarge)
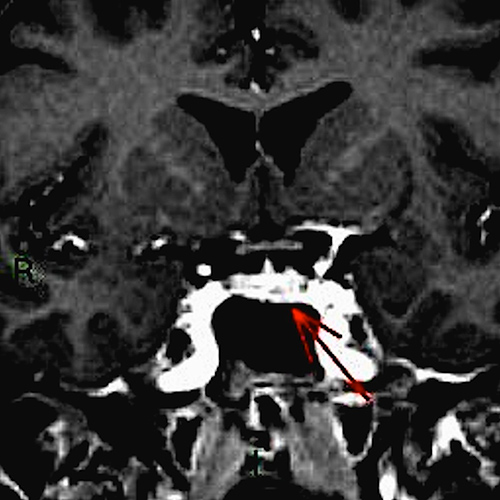
Figure 2 (click to enlarge)
Figure 1: Coronal contrast-enhanced T1 head MRI.
Figure 2: Coronal contrast-enhanced T1 head MRI; the arrows indicate the suspected micro-adenoma.
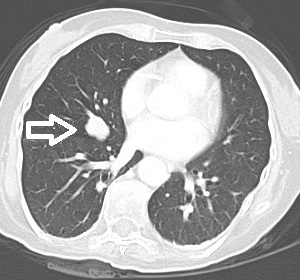
Figure 3 (click to enlarge)
Figure 3: Axial contrast enhanced chest CT; the arrows indicate the carcinoid tumour.
A right lower lobectomy was performed to remove the mass and histology revealed a typical carcinoid tumour. Immunohistochemical examination showed strong and diffuse positive staining for chromogranin and ACTH. Shortly after removal of the bronchial mass, blood pressure, serum potassium and blood glucose levels returned steadily to within normal ranges. Gastrin, calcitonin, PTH and chromogranin retesting showed their normalization. ACTH and cortisol levels gradually declined to subnormal ranges in the first week after surgery. The patient’s overall clinical condition gradually improved until discharge from hospital.
DISCUSSION
In the specific case of suspected pituitary ACTH-dependent CS, radiological imaging can be misleading in both a false positive and a false negative way. In this case, superficial interpretation of partial diagnostic data could have led to a hasty and incorrect intervention. In fact, the finding of a pituitary micro-adenoma initially reinforced a diagnosis of Cushing'd disease (CD), while the negative response to hormonal tests (CRH/desmopressin stimulation and IPS testing) indicated an ectopic ACTH source. Exclusion of a false neuroendocrine cause is mandatory as it has a bad prognosis, and in this case correct interpretation of diagnostic results avoided an unnecessary surgical approach. CS can be related to a wide variety of tumours. A newly discovered pituitary adenoma might be presumed to be the original cause of this syndrome; however, an ectopic ACTH-secreting tumour should always be excluded, as these tumours account for about 15% of all cases. Therefore, a neuroradiological finding of a pituitary adenoma should be followed by a thorax imaging to rule out an ACTH source other than pituitary . Stimulation tests and imaging studies are helpful but have limitations. CRH/desmopressin and IPS testing both have high specificity for extra-pituitary ACTH-secreting tumours, but some tumours might respond to the first test, while the absence of a gradient in the second does not exclude CD CS[6]. Due to poor MRI resolution, pituitary imaging often fails to detect adenomas as they can be very small, as is normally the case for most ACTH-secreting micro-adenomas. In addition, a positive result is not pathognomonic of the disease as incidental adenomas can be found in 10% of the general population[7].
A 6 mm tumour size cut-off has been recently suggested as the optimal threshold providing high specificity (96%), adequate to support a pituitary origin of CS and avoid IPS sampling in the diagnostic pathway[8]. The rationale for this cut-off comes from the evidence that pituitary incidental tumours rarely exceed 6 mm; in contrast, an adenoma <6 mm suggests EAS. Despite the high specificity of such a cut-off, there is a significant overlap in tumour size between ACTH-secreting and non-functioning adenomas. Consequently, new dynamic high-resolution MRI equipment is welcome as it can help to differentiate pituitary and ectopic aetiology, although an increase in incidental micro-adenoma diagnoses can be expected.
Given the limitations of dynamic tests and MRI, careful clinical evaluation of the patient is warranted as it can help the clinician to follow the correct diagnostic process. Clinical manifestations of EAS can be somewhat peculiar, as they frequently include features like psychiatric symptoms, severe hypertension and hypokalaemia whose onset and worsening are usually faster than in CD. In contrast, time to diagnosis in CD can be several months due to the gradual presentation of symptoms[9] which are the expression of a chronic exposure to cortisol excess.
CONCLUSION
Diagnosing ACTH-dependent CS can be difficult. Basic hormonal and imaging data may be insufficient to reach an appropriate diagnosis. Therefore, we emphasize the value of properly interpreting the clinical features and history of the cortisol excess, as this can help in discriminating between pituitary and ectopic CS. It is highly recommended that all patients be assessed jointly by a team of internists, endocrinologists, neurosurgeons and radiologists, as each can shed light on the ‘blind spots’ in the clinical experience of their colleagues.