ABSTRACT
A 76-year-old man was admitted to hospital with fever, weight loss, pancytopenia, hepatosplenomegalyand a double monoclonal component IgM-IgG-k, suggesting a diagnosis of myeloma. Bone marrow and liver biopsies disclosed the presence of Donovan bodies, andthe titre of anti-Leishmania antibodies was extremely high. After treatment with liposomal amphotericin B,the titre of antibodies fell considerably, while monoclonal components, pancytopenia and clinical symptoms slowly disappeared. Polyclonal γ-globulinsare made of innumerable monoclonal components, one of which can appear as a recognizable band and be misdiagnosed as myeloma when representing the hightitre of an antibody directed towards a specific antigen
LEARNING POINTS
- Rare diseases, although rare, do occur, even when not included in the differential diagnosis.
- The differential diagnosis of fever, leucopoenia and liver/spleen enlargement includesleishmaniasis.
- A double IgM-IgG monoclonal antibody should raise suspicion of a recent severe infection.
- There are cases of apparent liver cirrhosis, with highly suggestive elastometry stiffness values,which are reversible.
KEYWORDS
Leishmaniasis, multiple myeloma, lymphoma, liver cirrhosis, M-components
INTRODUCTION
The appearance of monoclonal components in serum focuses attention on multiplemyeloma or lymphoma. Pancytopenia associated to spleen/liver enlargement has a broad differential diagnosis, further complicated by its association with adouble monoclonal component. We report herein a patient exhibiting these features, in whom the diagnosis could have been missed or delayed ifpancytopenia, unreported in the area where the patient was living, had not been taken into consideration, and the M-components had not been linked to thepresence of a high titre of antibodies caused by the offending agent.
CASE DESCRIPTION
A male Caucasian patient, aged 76, was admitted to an Internal Medicine ward inMarch 2009 because of worsening dyspnoea, fatigue, and ankle oedema.
Thereferral diagnosis was monoclonal gammopathy of undetermined significance (MGUS) with double IgG-IgM-k components, increased γ-globulins and refractorypancytopenia with multi-linear dysplasia (Table 1).
| 5690 mg/dL | |
| IgG | 362 mg/dL |
| IgA | 210 mg/dL |
| P k | 1400 mg/dL |
| λ P | 525 mg/dL |
| U k | 42.2 mg/dL |
| U λ | 27.8 mg/dL |
| P β2 microglobulin | 16,578 ng/mL |
| U β2 microglobulin | 21,565 ng/mL |
| Daily proteinuria | 417 mg |
| INR | 1.32 |
| aPTT | 46.8 s |
| p 14 | + |
| p 16 | + |
| Ab anti- Leishmania | 1/5120 |
Table 1 - Data obtained on patient's admission, referring to measurements indicated by the international nomenclature, as explained in the text.
The physical examination disclosed jugular vein swelling, increased central venouspressure (CVP), oedema of the lower limbs, marked painless liver and spleen enlargement, a few spider nevi on his face and no palmar erythema or flappingtremor. Chest, heart and neurologic examinations were normal. His fever spiked daily up to 38°C. Although repeated blood cultures were negative, he receivedceftazidime, amikacin, darbepoietin and granulocyte colony stimulating factor (GCS-F). Results of the critical blood exams are reported in Table 1.
The bone marrow biopsy showed increased cellularity, and marked interstitial andperivascular plasma cell infiltration, positive to both κ and λ chains, organized in small clusters and amounting to 35% of all marrow cells. There wasevidence of dyserythropoiesis, occasional micromegakaryocytes and an important fibrotic argyrophilic reticulum.
Thesefindings were interpreted as suggestive of myelodysplasia associated toplasmacytosis and marrow fibrosis (Fig. 1).
Nuclearmagnetic resonance (NMR) disclosed increased liver size, left lobe hypertrophy, portal vein of 20 mm diameter and spleen of 20×20 cm.
Liverelastometry measured a stiffness of 27.7 KPa, compatible with liver cirrhosis.
Liverbiopsy evidenced pericellular and perisinusoidal fibrosis, with regenerative nodules, and a polymorphous portal and periportal inflammatory infiltrate,formed by granulocytes and macrophages (Fig. 2).
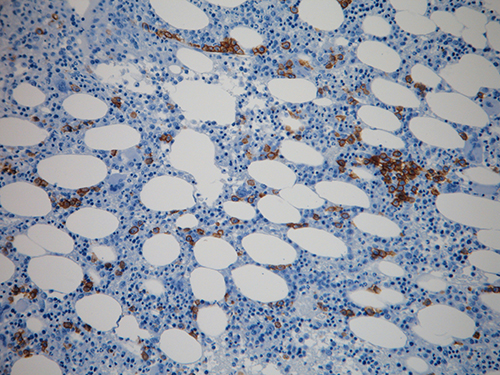
Figure 1 (click to enlarge)
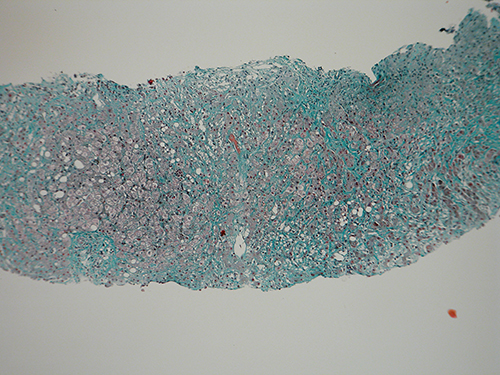
Figure 2 (click to enlarge)
Fig. 1 - Bone biopsy. The interstitial plasmocytosis is shown by the histochemical reaction with anti-CD138 antibody, revealed by the rusty colour (magnification 20×, DAB/haematoxylin stain). For explanation see text.
Fig. 2 - Liver biopsy. Active septae are evident, subdividing nodules with peripheral necrosis and cell ballooning. One nodule has been invaded by fibrotic tissue isolating single cells/groups of cells. The sinusoidal and pericellular fibrosis with active septae is suggestive of chronic active hepatitis evolving towards cirrhosis (magnification 10×, Trichromic Masson stain).
At higher magnification, numerous Donovan bodies were recognized inside liver cells and macrophages (Fig. 3).The bone marrow biopsy, re-examined, also showed Donovan bodies inside histiocytes (Fig. 4).
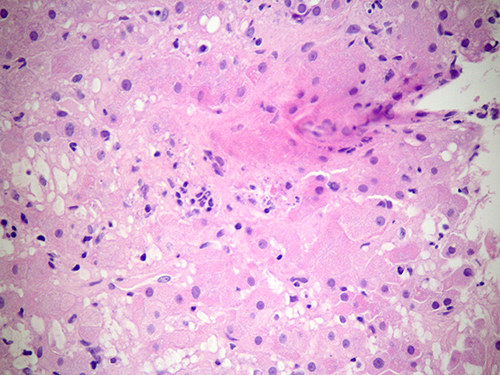
Figure 3 (click to enlarge)
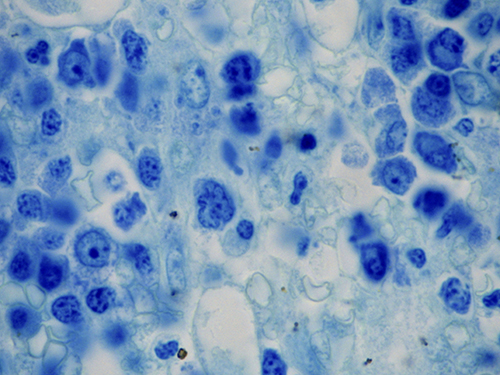
Figure 4 (click to enlarge)
Fig. 3 - Liver biopsy. Ballooned liver cells and macrophages contain several Donovan bodies in their cytoplasm (100×, Giemsa).
Fig. 4 - Bone marrow biopsy. Rare macrophages and/or reticulum cells contain several Donovan bodies in their cytoplasm (100×, Giemsa).
The titre of anti-Leishmania antibodies was executed at the “Istituto Superiore di Sanità”, Rome (Italy), and was foundpositive at a 1/5120 dilution, associated to a strong positivity to p14 and weak positivity to p16 by Western blot. Leishmaniasis is unreported inresidents in Piedmont (Italy). However, the patient spent the summer months in the county of Savona (Liguria, Italy), where an epidemic source of infection isknown and Phlebotomus papatasi, a vector insect of Leishmania, is present along the coast. The patient was treated with liposomal amphotericin B(Ambisome) 5 mg/kg/day for 5 consecutive days and then once weekly for 5 weeks.
Thedisease remitted completely: IgG fell to 1430, IgM to 115, IgA to 220 mg/dl and antibody titre to 1/2520. The hepatosplenomegaly reverted to normal, and so didthe abnormal liver exams, while pancytopenia disappeared.
DISCUSSION
The differential diagnosis of this patient was difficult, as several entities arecharacterized by hepatosplenomegaly, pancytopenia fever and a double IgM-IgG monoclonal component. Felty’s syndrome was unlikely because of the absence of rheumatoidarthritis[1], while a T-hepatosplenic lymphoma, although rare, was possible. However, this and angioimmunoblastic lymphoma are aggressive, leading to aprogressive disruption of liver structure, attended by jaundice and ascites. In contrast, the present patient displayed a more chronic course, more typical ofT-large granular lymphocytes leukaemia, a disease associated with rheumatoid arthritis and red cell aplasia, which were not present in this case. Hairy cellleukaemia (HCL) fitted the picture, but hairy cells were not found in his peripheral blood. The monoclonal components were suggestive of other B-celllymphomas, namely prolymphocytic leukaemia, because of liver/spleen involvement. However, the typical plasmacytoid lymphocytes were not observed in the patient’speripheral blood.
HepatosplenicT-cell lymphoma (HSTL) is a neoplasm of mature T cells that infiltrates the sinusoids of the spleen, liver and bone marrow[2]. Most patients exhibit markedhepatosplenomegaly without lymphadenopathy at physical examination, and the blood exams demonstrate thrombocytopenia. Other common findings includesystemic B symptoms (weight loss, fever, night sweats), anaemia, neutropenia and abnormal LFTs. The diagnosis is based on the accumulation of malignant atypicallymphocytes expressing CD2/CD3/CD7/CD16, γ/δ-T-cell receptors and/or α/β-T-cell-receptors. This condition was ruled out because of the absence ofthe typical histological pattern.
Angio-immunoblasticT-cell lymphoma (AITL) is one of the more common peripheral T-cell lymphomas and is thought to come from peripheral CD4-positive T cells, perhapscorresponding to a subset of follicular helper T-cells. Patients typically present with acute onset of generalized lymphadenopathy, hepatosplenomegaly andsystemic B symptoms with or without a rash, occasionally associated with autoimmune haemolytic anaemia, plasmacytosis or polyclonalhypergammaglobulinaemia. The diagnosis of AITL is best made by excisional biopsy of a lymph node[3]. These two last entities are aggressive, leading to aprogressive disruption of liver structure, attended by jaundice and ascites, whereas the present patient displayed a more chronic course.
T-largegranular lymphocyte (LGL) leukaemia is a clonal disease of the large granular lymphocyte characterized by peripheral blood and marrow lymphocytic infiltration withLGLs, splenomegaly and neutropenia. LGL leukaemia can be of T- or NK-cell lineage[4]. Even though monoclonal gammopathy of undetermined significance(MGUS) and multiple myeloma have been described in association with LGL leukaemia, this disease was ruled out because it requires association withrheumatoid arthritis and red cell aplasia.
HCLcauses splenomegaly (which may be massive), systemic complaints, bruising and bleeding secondary to severe thrombocytopenia, and recurrent infectionssecondary to granulocytopenia and monocytopenia. A diagnosis of HCL is confirmed when the abnormal cells display pan-B cell antigens (e.g.CD19/CD20/CD22) along with positivity for CD103/CD11c/CD25[5]. The clinical features HCL fitted our patient, but hairy cells were not found in hisperipheral blood.
Prolymphocyticleukaemia (B-PLL) is a rare B-cell neoplasm comprising prolymphocytes, typically with involvement of the peripheral blood, bone marrow and spleen. It is most common in elderly Caucasians. A T-cell variant has been reported[2].
Falciparummalaria, nowadays unheard of in Italy in people who have not travelled to endemic areas, was ruled out by repeated peripheral blood smears. Mycobacteriumavium complex is being reported in HIV-positive patients[6], but the patient was negative. Typhoid fever was ruled out by the negative serum reaction andthe absence of roseola. Brucellosis causes leukocytosis[7], whereas the patient was leukopenic. Peripheral leukocytosis could have been expected also in portalvein phlebitis or abscess formation in the context of liver cirrhosis[8]. Having ruled out all the above possibilities, biclonal multiple myeloma was theonly possibility left, although this constitutes only 1% of all myelomas[9]. However, the progressive liver disease and worsening of clinical conditions,pancytopenia, spleen enlargement, fever with marked sweating and clinical signs suggestive of a systemic infection called for a unifying infectious hypothesis.Lieshmaniasis, though unheard of in this geographic area, did satisfy the clinical/laboratory picture, with the exception of the monoclonal bands, unlessthese were due to a markedly high titre of anti-Leishmania antibodies, detected during the switch from IgM to IgG.
Inconclusion, although leishmaniasis in northern Italy is increasingly reported only in travellers from endemic areas and countries[10], and is thus notusually suspected, it was indeed the correct diagnosis in this patient. The monoclonal bands, a “red herring” in the differential diagnosis, were in factdue to the high titre of specific anti-Leishmania antibodies, “frozen” in the blood sample during the switch from IgM to IgG.
Thiscase demonstrates how an apparently clear diagnosis of B-cell lymphoma (including initial multiple myeloma or Waldenström’s disease) and/or livercirrhosis could mimic a reversible condition.
Afterall, “polyclonal” γ-globulins are made of an almost unlimited number of small monoclonal components, each specific for an offending agent. It should come asno surprise that, during the acute antibody response to an antigen, the monoclonal component could be detected, suggesting multiple myeloma orlymphoma. This M-component should disappear with the drop in antibody titre, as happened in our patient. In fact, a similar case due to Babesia infection has been previously reported, suggesting that protozoan infections may be more likely to cause pseudo-myeloma or -lymphoma.