ABSTRACT
Immune-mediated necrotizing myopathies (IMNM) are recognized as a subgroup of idiopathic inflammatory myopathies (IIM). IMNM are defined based on a combination of clinical presentation and laboratory studies, requiring a specific myopathological pattern on muscle biopsy for diagnosis. The authors describe a case of a patient with necrotizing myopathy, thought to be immune mediated, highlighting the challenge of its differential diagnosis. As clinical assessment and diagnostic tools sometimes fail to determine whether a necrotizing myopathy is immune mediated, leading to misdiagnosis and a compromise of the optimal therapeutic approach, distinguishing between IMNM and other myopathies is crucial.
LEARNING POINTS
- IMNM are recognized as a subgroup of idiopathic inflammatory myopathies along with dermatomyositis, polymyositis, inclusion body myositis and non-specific myositis.
- IMNM diagnosis is based on clinical features, laboratory studies (serum CK levels,electromyography, serum myositis-specific autoantibodies) and myopathological features. Prominent muscle fibre necrosis and regeneration with minimal inflammatory infiltrate is the predominant abnormal histological feature.
- As clinical assessment and diagnostic tools sometimes fail to determine whether a necrotizing myopathy is immune mediated, leading to misdiagnosis and a compromise ofthe optimal therapeutic approach, distinguishing between IMNM and other myopathies is crucial. When there is no clear demonstration of exogenous toxic contact and no other known aetiologies can be supported by evidence,even in the absence of autoantibody seropositivity, an immunosuppressive treatment should be attempted based on the hypothesis of an immune-mediated myopathic injury.
KEYWORDS
Myopathy, necrotizing myopathy, immune-mediated necrotizing myopathy
INTRODUCTION
Myopathy defines a muscle disease that can be inherited or acquired. Idiopathic inflammatory myopathies (IIM) are a heterogeneous set of acquired muscle disorders. Distinct categories have been proposed based on a combination of clinical presentation, laboratory studies and pathological findings in muscle biopsy: polymyositis, dermatomyositis, inclusion body myositis (IBM), non-specific myositis (NSM) and immune-mediated necrotizing myopathies (IMNM)[1]. The last subgroup constitutes a disorder with necrotic myofibres without significant inflammation as the predominant abnormal histological feature[1]. Necrotizing myopathies can also be secondary to drugs or associated with connective tissue disorders or cancer[2].
The authors describe a case of a patient with necrotizing myopathy, thought to be immune mediated, highlighting the challenge of its differential diagnosis.
CASE PRESENTATION
A 66-year-old woman presented with a 12-month history of progressive symmetrical proximal muscle weakness and discrete myalgia of the upper limbs. She complained of fatigue with progressive disability for daily routine activities. Mild dysphagia was initially present, but did not persist. She reported none of the following: dermatologic alterations; arthralgia; weight loss; eye, mouth and respiratory symptoms. The patient had a history of inguinal Hodgkin lymphoma (mixed cellularity type, stage IIA) treated with chemo- and radiotherapy and had been in complete remission for 18 years. She reported no regular medication, including lipid-lowering drugs, or toxic contact. No relevant family history was reported. Physical examination showed proximal weakness in the arms (Medical Research Council grade 3/5 for elbow flexion and extension and shoulder abduction) and legs (grade 4/5 for hip and knee flexion and extension); sensory and skin examination was normal.
Laboratory tests showed elevated levels of creatine kinase (CK) (3509 IU/l), myoglobin (829 ng/ml), aldolase (47.2 IU/l), MB-CK (125.7 ng/ml) and lactate dehydrogenase (588 IU/l). Serum aminotransferase levels were elevated (aspartate transaminase/alanine transaminase 81/133 IU/l) and troponin I levels were normal. Serum creatinine was decreased (0.37 mg/dl); erythrocyte sedimentation rate (17 mm/h) and C-reactive protein CRP (0.10 mg/dl) were normal. No haemogram alterations or electrolyte imbalance were found. Thyroid and parathyroid hormones, thyroid autoantibodies, HbA1c, complement, serum immunoglobulins and protein electrophoresis were normal. Testing for viral and bacterial antibodies was negative (HIV, hepatitis B and C, herpes, Epstein–Barr virus, cytomegalovirus, Treponema pallidum, Mycoplasma pneumoniae, zoonotic bacteria). Antinuclear antibodies (ANA) were moderately positive with a fine dense granular pattern (1/160). Immunodiagnostic tests for myositis-associated autoantibodies (MAA) and myositis-specific autoantibodies (MSA) were all negative (Table 1) Electromyography showed spontaneous activity (fibrillation potentials and positive sharp waves, complex repetitive discharges) in proximal muscles, consistent with irritable myopathy. Nerve conduction was normal. Nail capillaroscopy, urine testing, oesophageal manometry, upper endoscopy, echocardiogram, functional respiratory testing and thoracic-abdominal-pelvic CT scan showed no relevant alterations.
| Autoantibody testing | Result |
|---|---|
| Myositis-associated autoantibodies (MAA) | |
| Anti-PM-Scl | negative |
| Anti-RNP | negative |
| Anti-Ku | negative |
| Anti-Ro-60 | negative |
| Myositis-specific autoantibodies (MSA) | |
| Antisynthetase autoantibodies | |
| Anti-JO-1 | negative |
| Anti-PL-7 | negative |
| Anti-PL-12 | negative |
| Anti-EJ | negative |
| Anti-OJ | negative |
| Nonsynthetase autoantibodies | |
| Anti-SRP (anti-signal recognition particle) | negative |
| Anti-Mi-2 | negative |
| Anti-MDA5 | negative |
| Anti-155/140 | negative |
| Anti-SAE | negative |
| Anti-NXP2 | negative |
| Anti-HMGCR (anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase) | not available for routine testing |
| Other autoantibodies | |
| All ANA (antinuclear autoantibodies) | moderately positive with a fine dense granular pattern (titre: 1/160) |
| Anti-ds-DNA | negative |
| Anti-SS-B | negative |
| Anti-Sm | negative |
| Anti-Scl70 | negative |
| Anti-ANCA (anti-neutrophil cytoplasmic autoantibodies) | negative |
| Anti-thyroglobulin | negative |
| Anti-thyroid peroxidase | negative |
Table 1: Patient autoantibody testing
A left deltoid muscle biopsy was performed. Immunohistological examination demonstrated necrotic and regenerating muscle fibres (Figs. 1 and 2), discrete inflammatory infiltrate (Fig. 2), strong MHC-I expression on necrotic fibres and mild expression on non-necrotic ones (Fig. 3). It also showed preserved dysferlin expression and no perifascicular atrophy or vacuoles.
The patient was diagnosed with necrotizing myopathy and 60 mg/day of oral prednisolone (1 mg/kg/day) was started with a significant improvement in functional abilities and proximal muscle strength after 2 weeks. Corticosteroid doses were tapered over a year, with sustained clinical and analytical improvement. Nearly normal CK and aldolase levels were attained after 10 months of treatment.
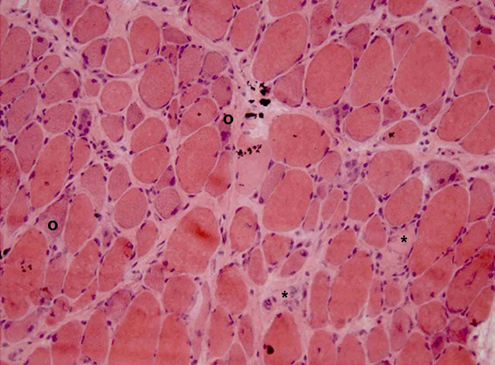
Figure 1 (click to enlarge)
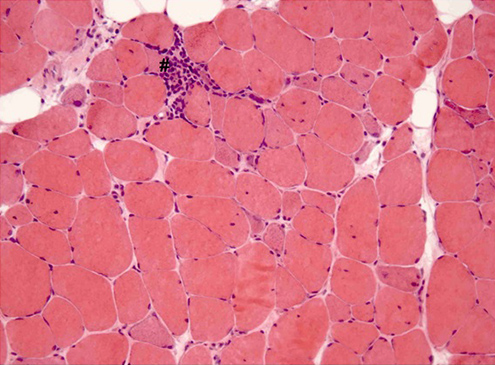
Figure 2 (click to enlarge)
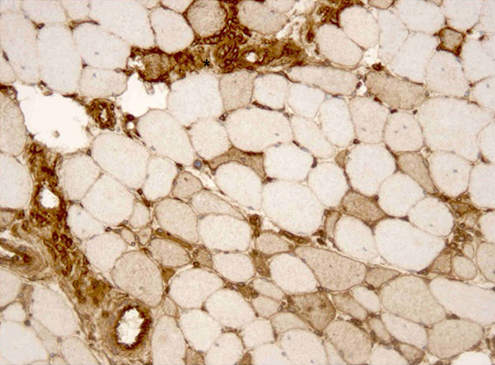
Figure 3 (click to enlarge)
Figure 1: Left deltoid muscle biopsy. Increased variability of muscle fibre diameter with necrotic fibres (º) and regenerating fibres with basophilic cytoplasm (*). Haematoxylin-eosin staining; ×200.
Figure 2: Left deltoid muscle biopsy. Necrotic and regenerating fibres, minimal inflammatory infiltrate (#). Hematoxylin-eosin staining; ×200.
Figure 3: Left deltoid muscle biopsy. Strong major histocompatibility complex (MHC-I) expression on necrotic fibres (*) and mild expression on some non-necrotic fibres. MHC-I immunoperoxidase staining, ×200.
DISCUSSION
IMNM are defined as ‘fulfilling clinical inclusion criteria of adult dermatomyositis or polymyositis, with subacute or insidious post-puberty onset of symmetric proximal > distal limb and neck flexor > neck extensor weakness, elevated serum CK levels and detection of MSA, as well as exclusion criteria as IBM clinical features, dermatomyositis typical rash, ocular weakness, isolated dysarthria and signs of toxic myopathy, endocrinopathy, family history of muscular dystrophy or proximal motor neuropathies’. Many necrotic muscle fibres are considered the predominant histological feature; inflammatory cells are sparse or slightly perivascular[1]. IMNM have been misclassified in the past as dermatomyositis or polymyositis[1].
As the patient presented muscle weakness and fatigue with elevated muscle enzymes, metabolic myopathies and systemic inflammatory disease-associated myopathies needed to be considered. The patient presented no joint, lung, heart or renal involvement, and the inflammatory serum markers, hormonal, electrolyte and infection laboratory parameters were normal. Despite the initial mild dysphagia, oesophageal manometry and upper endoscopy showed no alterations.
As the patient fulfilled the clinical and laboratory criteria for an IIM, the differential diagnosis had to rely on skin alterations and myopathological features[1]. Dermatomyositis was ruled out by the absence of skin lesions, perifascicular atrophy and significant perivascular inflammation. The pathological findings were also distinct from that of polymyositis, as there was no significant endomysial infiltrate or ubiquitous MHC-I expression. The absence of rimmed vacuoles and ragged red fibres excluded IBM[1].
Some necrotizing myopathies can be drug induced[2]. Although the patient presented clinicopathological features that may suggest a toxic myopathy, there was no epidemiological context. There was no evidence of exogenous myotoxic exposure, except when she was on Hodgkin lymphoma treatment, 18 years prior to myopathy onset. The time gap was too wide to establish a cause–effect association, thus ruling out a drug-induced aetiology.
Muscle fibre necrosis and regeneration with sparse inflammation is also observed in muscular dystrophy, but the non-prolonged clinical evolution, the absence of a family history of myopathy, the preserved dysferlin expression and the early response to immunosuppressants ruled out the hypothesis of muscular dystrophy[2].
Paraneoplastic necrotizing myopathies share clinical, laboratory and pathological features with IMNM[2]. An underlying malignancy was excluded by the patient’s laboratory and diagnostic imaging evaluation, ruling out a paraneoplastic framework.
Although many IMNM pathophysiological mechanisms are not entirely understood, the MSA identification suggests an antibody-mediated disease, in which antibodies may recruit cytotoxic immune effector cells via antibody-dependent cell-mediated cytotoxicity. As the prominent histopathological feature of IMNM, necrosis translates a cytotoxic process. Whether IMNM are directly triggered by unrecognized endogenous myotoxics or they are antibody-mediated diseases remains unclear[3].
Some MSA have been associated with IMNM: anti-SRP and anti-HMGCR[2,4,5]. Usually, IMNM with positive anti-SRP autoantibodies has rapidly progressive disabling muscle weakness and is frequently associated with organ involvement[2,4]. Anti-HMGCR autoantibodies have been identified in IMNM patients without a history of statin exposure[5]. The patient was not tested for anti-HMGCR, as testing was not available.
It has been reported that about 50% of the IIM patients are autoantibody seronegative[6], implying that an antibody-mediated process is not an underlying pathophysiological mechanism of the disease. Although the patient presented moderately positive ANA, these antibodies may be positive in healthy individuals, lacking diagnostic power for autoimmune diseases. Despite the negative MAA and MSA testing, an immune-mediated myopathic injury was hypothesized for this patient and immunosuppressive treatment was considered suitable according to this hypothesis. There was an effective therapeutic response, with clinical and analytical improvement after corticosteroid treatment, strongly supporting the IMNM hypothesis.
Clinical assessment and diagnostic tools sometimes fail to determine whether a necrotizing myopathy is immune mediated and may lead to misdiagnosis and a compromise of the optimal therapeutic approach.